Adenylosuccinate Lyase Deficiency (dADSL)
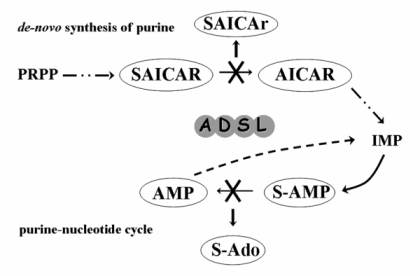
The adenylosuccinate lyase deficiency (MIM# 103050) is an inherited autosomal recessive disorder of the purine metabolism. There is an enzyme adenylosuccinate lyase (ADSL; EC 4.3.2.2) affected in this disease. This enzyme catalyses the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) into aminoimidazole carboxamide ribotide (AICAR) in the de novo purine synthesis pathway, and the formation of adenosine monophosphate (AMP) from adenylosuccinate (S-AMP) in the purine nucleotide cycle (Fig. 1) [1, 2].
ADSL functions in a form of a homotetrametric complex under native conditions. The ADSL subunits are encoded in humans by a single ADSL gene (MIM# 608222), which spans 23 kb on chromosome 22q13.1-13.2. The gene consists of 13 exons, and it is ubiquitously expressed as two alternatively spliced mRNA isoforms [3-5].
The ADSL deficiency leads to the serious neurological symptoms. The rate of the disablement is various and the disease can be clinically classified as three basic types: neonatal form (prenatal hyperkinesis, lung hypoplasis, and prenatal abortion of growth, following by fatal neonatal encephalopathy) [6], severe infant form – type I (heavy psychomotor retardation, frequently early death) [7, 8], moderate to mild form – type II (psychomotor retardation, hypotony and autism) [7, 9, 10].
The pathogenic mechanism(s) leading to the development of individual clinical symptoms and underlying the phenotypic heterogeneity remain(s) unclear. The main pathogenic effect has been attributed to the toxic effects of accumulating dephosphorylated substrates of adenylosuccinate lyase – of the succinyladenosine (S-Ado) and of the succinylaminoimidazolecarboxamide riboside (SAICAr) in body fluids [11]. Although the absolute S-Ado and SAICAr concentrations do not correlate with the severity of the phenotype, it has been found that different values for the ratio between concentrations of S-Ado and SAICAr in the cerebrospinal fluid (S-Ado/SAICAr ratio) do correspond with the three main phenotypic groups [6, 12].
Biochemical diagnostics of the disease is based on the detection of S-Ado and SAICAr in the urine, plasma and cerebrospinal fluid and the molecular biology diagnostics on the detection of the mutations in the ADSL gene.
The diagnostics of the patients with the adenylosuccinate lyase deficiency is established from 1993 in our laboratory (IIMD Czech Republic). It is based on the investigation of children with non specific psychomotoric disorders, enzymatic examination of biochemical positive patients, analysis of the mutations of the gene for ADSL and verification of the pathogenity of the private mutations [4, 6, 13-17]. We provide a complex diagnostics to clinical sites not only in Czech Republic, but also abroad via the GeneTests system. We also offer commercially unavailable substrates of adenylosuccinate lyase – SAdo, SAICAr and SAICAR – for diagnostics and research [18]. For more information please contact marie.zikanova@lf1.cuni.cz.
To date, about 70 patients with ADSL deficiency and 49 different ADSL mutations have been reported. A summary of all of them you can see in Database.
[1] Ciardo, F., C. Salerno, and P. Curatolo, Neurologic aspects of adenylosuccinate lyase deficiency. J Child Neurol, 2001. 16(5): p. 301-8.
[2] Jaeken, J. and G. Van den Berghe, An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet, 1984. 2(8411): p. 1058-61.
[3] Fon, E.A., et al., Mapping of the human adenylosuccinate lyase (ADSL) gene to chromosome 22q13.1-->q13.2. Cytogenet Cell Genet, 1993. 64(3-4): p. 201-3.
[4] Kmoch, S., et al., Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum Mol Genet, 2000. 9(10): p. 1501-13.
[5] Van Keuren, M.L., et al., A somatic cell hybrid with a single human chromosome 22 corrects the defect in the CHO mutant (Ade-I) lacking adenylosuccinase activity. Cytogenet Cell Genet, 1987. 44(2-3): p. 142-7.
[6] Mouchegh, K., et al., Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: observation of 6 patients in 4 families. J Pediatr, 2007. 150(1): p. 57-61 e2.
[7] Jaeken, J., et al., Adenylosuccinase deficiency: an inborn error of purine nucleotide synthesis. Eur J Pediatr, 1988. 148(2): p. 126-31.
[8] Maaswinkel-Mooij, P.D., et al., Adenylosuccinase deficiency presenting with epilepsy in early infancy. J Inherit Metab Dis, 1997. 20(4): p. 606-7.
[9] Jaeken, J., et al., Adenylosuccinase deficiency: a newly recognized variant. J Inherit Metab Dis, 1992. 15(3): p. 416-8.
[10] Valik, D., P.T. Miner, and J.D. Jones, First U.S. case of adenylosuccinate lyase deficiency with severe hypotonia. Pediatr Neurol, 1997. 16(3): p. 252-5.
[11] Stone, T.W., et al., Succinylpurines induce neuronal damage in the rat brain. Adv Exp Med Biol, 1998. 431: p. 185-9.
[12] Van den Bergh, F., et al., Residual adenylosuccinase activities in fibroblasts of adenylosuccinase- deficient children: parallel deficiency with adenylosuccinate and succinyl-AICAR in profoundly retarded patients and non-parallel deficiency in a mildly retarded girl. J Inherit Metab Dis, 1993. 16(2): p. 415-24.
[13] Jurecka, A., et al., Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol Genet Metab, 2008. 94(4): p. 435-42.
[14. Krijt, J., et al., An unique case of false-negative screening for ADSL deficiency in urine possibly caused by bacterial infection. Clin Chem, 2010(in preparation).
[15] Krijt, J., et al., Identification and determination of succinyladenosine in human cerebrospinal fluid. J Chromatogr B Biomed Sci Appl, 1999. 726(1-2): p. 53-8.
[16] Sebesta, I., et al., Adenylosuccinase deficiency: Clinical and biochemical findings in 5 Czech patients. Journal of Inherited Metabolic Disease, 1997. 20(3): p. 343-344.
[17] Zikanova, M., et al., Biochemical and structural analysis of 14 mutant ADSL enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum Mutat, 2010. 31(4): p. 445-55.
[18] Zikanova, M., et al., Preparation of 5-amino-4-imidazole-N-succinocarboxamide ribotide, 5-amino-4-imidazole-N-succinocarboxamide riboside and succinyladenosine, compounds usable in diagnosis and research of adenylosuccinate lyase deficiency. J Inherit Metab Dis, 2005. 28(4): p. 493-9.