Použité zkratky:
AβP - amyloid beta protein; AEF - amyloid enhancing factor; AH - amyloid heavy chain; AL - amyloid light chain; ANP - atrial natriuretic protein; ApoA1/2/4 (ApoAI/II/IV) -apoliprotein A; APP - amyloid precursor protein; β2MG - β-2-microglobulin; Bri - Bri protein; Cal - calcitonin; CJD - Creutzfeldt-Jakob disease; Cys - cystatin; ECM - extracellular matrix; FAD - familial Alzheimer´s disease; FAP - familial amyloid polyneuropathy; FFI - Fatal Familial Insomnia; Fib - fibrinogen; GAG - glykosaminoglykany; Gel - gelsolin; GPI - glycosyl-phosphatidylinositol; GSS - Gerstmann-Sträussler-Scheinker disease; IAPP - islet amyloid precursor protein; Ins - insulin; Ker -keratoepithelin; Lac - lactoferrin; Lys - lysozyme; m.w. - molecular weight;
Med - medin; MHC - main histocompatibility complex; MIDD - monoclonal imunoglobulin deposition disease; NFT - neurofibrillary tangles; OMIM - Online Mendelian Inheritance in Man; PHF - paired helical filaments; PrP - Prion protein; PS 1/2 - presenilin 1/2; SAA - serum amyloid A protein; SAP - serum amyloid P component; Sgl - semenogelin; TGF β-1 - transforming growth factor β-1; TTR - transthyretin
Amyloid je patologická fibrilární forma proteinu v beta struktuře (struktuře skládaného listu). Ke vzniku amyloidu dochází agregací původně solubilní (degradovatelné) formy proteinu s primárně nebo sekundárně výrazně zastoupenou beta strukturou do formy fibrilární (resistentní na degradaci).
Polypeptid složený do struktury skládaného listu je v extendované neboli rozvinuté či napřímené konformaci. Stabilizace nastává především interakcí mezi jednotlivými polypeptidickými řetězci. Dochází tak , na rozdíl od alfa helixu, kde stabilizace probíhá uvnitř šroubovice, nejen k intramolekulární interakci mezi vzdálenými částmi polypeptidického řetězce, ale i k i intermolekulární agregaci mezi jednotlivými proteiny.
Vzniklé fibrily se skládají z beta úseků kde jednotlivé řetězce jsou navzájem uspořádány antiparalelně a leží kolmo na dlouhou osu vlákna. Od nich je odvozen alternativní název amyloidos - beta fibrilosy. Existuje řada pozorování svědčící pro existenci protofibril (viz dále). Fibrily jsou velmi resistentní na proteolysu a jejich degradace je tedy velmi omezená (viz níže).
Mechanismus vzniku amyloiduPodmínkou vzniku amyloidu je narušený životní cyklus proteinu se vznikem abnormálního přechodného mezičlánku s beta strukturou, náchylného k fibrilární agregaci. Podle současných představ vzniká tento amyloidogenní mezičlánek buď ve fázi vzniku nebo ve fázi degradace amyloidogenního proteinu. V dnešním pohledu je přítomnost beta struktury (varianta sekundární struktury) pro vznik amyloidní fibrily esenciální. Podle toho existují proteiny primárně náchylné za určitých podmínek k fibrilární agregaci (viz níže), zatímco jiné proteiny se stávají amyloidogenními až po indukci beta konformace (viz priony). Agregace proteinu do amyloidní fibrily je tedy fyzikální proces, vznikající jako následek různých poruch v životním cyklu proteinu a to od stavu jeho zrodu do fáze jeho degradace a který odráží jeho abnormální konformaci. Amyloidosy lze tedy definovat jako abnormální biologické stavy vedoucí k optimálním podmínkám pro spuštění zmíněného fyzikálního procesu vytváření amyloidních fibril. Tyto poruchy se týkají vždy jednoho proteinu. Jejich povaha je doposud známa jen částečně. Mutace amyloidogenního proteinu proces vzniku amyloidu významně urychluje.
Z dosavadních pozorování vyplývá, že může jít na jedné straně o poruchy v procesu sekrece, ať již látky standardně secernované exocytosou viz např. atriální natriuretický hormon, kalcitonin a další) nebo uvolnění amyloidogenního peptidu z buněčné membrány proteolytickým zásahem (ectodomain shedding) s jeho následnou fibrilární agregací převážně nebo výlučně v bezprostředním okolí sekreční buňky. Takto lze nahlížet např. na cerebrální amyloidy na basi APP (viz níže). V každém případě lze předpokládat, že v této skupině amyloidos jsou amyloidogenní proteiny secernované (pro možnou výjimku viz amyloid na basi cytokeratinu). Zákonitosti agregace do fibril jsou jen zčásti známé. Poněkud stranou stojí skupina amyloidos na basi prionů. Vznik amyloidu je zde vysvětlován změnou konformace alfa -> beta interakcí normálního prionu (alfa) s patologickým prionem (beta).
Dále je to veliká skupina amyloidos generalizovaných (systémových), u kterých dochází k fibrilární agregaci cirkulujícího secernovaného amyloidogenního proteinu (SAA, TRR, AL, lysozym, apoAI, fibrinogen, viz níže) v mnoha tkáních. V patogenese těchto stavů může hrát roli několik faktorů isolovaně nebo v kombinaci. Jednak jde často o zvýšenou koncentraci amyloidogenního proteinu v séru (viz dále SAA, beta2MG, AL), danou nedostatečným vylučováním amyloidogenního proteinu (viz beta2MG) nebo jeho hyperprodukcí (SAA, AL). Samotné zvýšení koncentrace amyloidogenního proteinu v extracelulární tekutině však není pravděpodobně hlavním faktorem. Velmi důležitou roli v molekulární patogenese generalizovaných amyloidos hraje zřejmě abnormální finální proteolysa amyloidogenního proteinu v tkáních, která vede doposud nejasným způsobem k optimálním podmínkám pro fibrilární agregaci celé molekuly amyloidogenního proteinu (nebo jeho fragmentů) v pericelulárním prostoru tkání a to ve značné vzdálenosti od buněk produkujících amyloidogenní protein nebo zodpovědných za jeho abnormální degradaci. To znamená, že oproti fysiologické (neamyloidogenní) cestě s předpokládanou kompletní degradací kritického amyloidogenního proteinu v lysosomech, je předpokladem vzniku systémové amyloidosy vznik bloku lysosomální degradace s nástupem alternativního mechanismu, jehož podstatu neznáme. Lze předpokládat, že by byla porucha omezena na daný amyloidogenní protein a že paralelní lysosomální katabolismus jiných proteinů probíhá nerušeně.
Teprve touto fibrilární agregací se stává porucha morfologicky postřehnutelnou. Pokud by se porucha, na př. defektní degradace spojená s regurgitací, týkala neamyloidogenního proteinu, jehož fragmenty neagregují do struktur vyššího řádu (amyloidních fibril, nebo jiných fibrilárních struktur), změny by probíhaly v morfologicky málo postřehnutelné nebo nepostřehnutelné formě (viz amyloid u Alzheimerovy nemoci).
Finální proteolytické mechanismy pro hlavní zmíněné amyloidogenní proteiny (viz níže) však nejsou dostatečně známé. Předpokládá se, že hlavním degradujícím elementem jsou zejména elementy mononukleárního fagocytárního systému. Nakolik jde o výlučnou intracelulární degradaci v endosomálním systému nebo zda k proteolyse může dojít též (nebo výlučně?) v extracelulárním prostoru (secernovanými proteasami?) není známo. Role lysosomálního systému v amyloidogenese všech typů je stále nejasná.
V patogenetickém řetězci, vedoucím k deposici amyloidu hraje důležitou roli faktor urychlující vznik amyloidu (amyloid enhancing factor: AEF). K objevu tohoto faktoru vedly klasické pokusy s vyvoláním generalisované amyloidosy opakovanými injekcemi kaseinu myším (indukce SAA, viz níže) kdy po iniciální, několikatýdenní lag fázi nastává fáze deposice amyloidu. Jde o biochemicky doposud nedokonale definovanou látku, připravenou extrakcí tkání postižených amyloidem (nezávisle na typu amyloidogenního proteinu). Je indukována v makrofázích. V tkáni se objevuje 24 - 48 hodin před deposicí amyloidu. Po intravenosní aplikaci AEF (nebo makrofágů z tkání pokusně vyvolávané amyloidosy) dochází k urychlené deposici amyloidu bez iniciální lag fáze. Tento faktor byl připraven i z tkání myší dědičně resistentních na indukci amyloidosy. Podle některých údajů je AEF glykoprotein o nízké molekulové hmotnosti s výrazným zastoupením beta struktury a významnou schopností asociace s řadou molekul. V experimentu měly tuto vlastnost pouze fragmentované fibrily amyloidu (testováno s SAA amyloidem), nikoliv testované průvodní komponenty amyloidu (SAP, GAG). Tyto amyloidní fibrily tak zřejmě představují nukleační centra vzniku amyloidu, která indukují precipitaci a fibrilární agregaci mechanismem přirovnávaným k prionosám. Pozoruhodné je, že vedle i.v. aplikace došlo k indukci amyloidosy i po podání zmíněného preparátu amyloidních fibril i při podání per os.
U všech shora uvedených faktorů nutno předpokládat významnou účast lokálních faktorů, určujících, kde k deposici amyloidu v tkáních dojde a jaký bude distribuční charakter amyloidosy. Ani při generalizovaných amyloidosách není nikdy generalizace úplná. Lze předpokládat, že místa deposice nemusí být identická s místy produkce amyloidogenních fragmentů. Rozdíly v postižení tkání mohou být značné, dokonce existují i rozdíly postižení uvnitř populace buněk stejného typu (např. adipocytů). Je všeobecně uznáváno, že jednou vzniklé ložisko amyloidu působí jako nukleační faktor (templát) pro další deposici (odpovídá to i shora uvedeným poznatkům o povaze AEF)
Obecnější zákonitosti lokalizace amyloidních deposit nejsou známé. Znalosti v této oblasti jsou pouze empirické. V minulosti byly amyloidosy rozlišovány podle toho, zda deposice začínala v oblasti basálních membrán a retikulárních vláken (t.zv. periretikulinový typ). Šlo o deposici v oblasti malých artérií, arteriol, sinusoid a žil, propagující se v dalším průběhu do parenchymu. Za významné bylo považováno postižení intimy a medie cév, glomerulárních kapilár. Z tehdy definovaných forem amyloidosy šlo o sekundární povšechnou amyloidosu (dnes SAA). Tak zvaný perikolagenní typ deposice byl charakterizován začátkem v adventicii cév (artérií i žil), v intersticiu ledvin (ne v glomerulech) a v oblasti sarkolemy srdečního a hladkého svalu a v nervech okolo Schwannových buněk. Sem byla řazena amyloidosa u mnohotného myelomu, t.zv. primární amyloidosa (dnes AL) a neuropatické formy. Toto dělení se dnes již nepoužívá vzhledem k značné variabilitě distribuce. Navíc je deposice i u perikolagenní formy ve významném vztahu i k basálním membránám uvedených lokalizací.
Další (koprecipitované) komponenty - nespecifické součásti amyloidních deposit.Mezi nejvýznamnější patří sérový amyloidní protein P (SAP), glykosaminoglykany, apolipoproteiny a některé další komponenty, deponující se nezávisle na typu amyloidu. Jako častá komponenta řady amyloidu byl prokázán ApoA1, což může způsobit rozpaky v diagnostice. Přítomnost koprecipitovaných komponent naznačuje, že vznik amyloidu je provázen celou řadou velmi významných dalších lokálních procesů indukovaných amyloidogenním procesem.
SAP je objemný komplex, skládající se ze dvou pentamerních jednotek. Jde o endogenní kalcium dependentní lektin, jehož nejlépe známými ligandy jsou beta-galaktosa a fosfoethanolamin (z dalších lze jmenovat DNA, fibronektin a glykosaminoglykany). SAP je normální komponentou ECM. Je přítomen v elastických vláknech a v glomerulární basální membráně. Jeho funkce v těchto lokalizacích není známá. Primární struktura SAP je do značné míry homologní s t.zv. C reaktivním proteinem. Podle současných znalostí je degradován pouze v játrech. Jeho poločas v plasmě je okolo 14 hodin.
Vazba iontu vápníku podmiňuje resistenci SAP na proteolytickou degradaci (která je možná jen v nepřítomnosti kalcia). Protože SAP je integrální součástí všech typů amyloidů (včetně cerebrálních u Alzheimerovy nemoci), přenáší zřejmě proteolytickou resistenci i na amyloid. Charakter SAP reaktivního ligandu amyloidu není znám. Rozvolnění této vazby je cílem moderních terapeutických studií. Vazba SAP na amyloid je možná nejen v průběhu fibrilární agregace, ale také dodatečně na preexistující fibrily amyloidu, čehož se využívá k diagnostice amyloidu in vivo (aplikace radioaktivně značeného SAP a sledování jeho vychytávání). SAP je v amyloidu v intaktní formě.
Experimentální amyloidosa (vyvolaná kaseinem) v myších s vyřazeným genem pro SAP probíhá pomaleji. Absence SAP nevedla sama o sobě k detekovatelných změnám.
Z glykosaminoglykanů je to zejména heparan sulfát (odpovídající perlecanu, typu deponovanému v extracelulární matrix, nikoliv na buněčné membráně, viz ). Jde o nekovalentní vazbu. Jejich kvantum je odhadováno na 1-2% hmoty amyloidu. Heparan sulfát byl prokázán ve všech typech amyloidu a to včetně amyloidů cerebrálních. Patogeneticky důležitou komponentou je pravděpodobně apolipoprotein E. V případě cerebrální neuronální amyloidosy na basi APP je přítomnost některých isoforem apoE (zejména apoE4) považována za významný risikový faktor rozvoje Alzheimerovy nemoci (viz níže). Z dalších komponent je to alfa1 antichymotrypsin. V amyloidních placích Alzheimerovy nemoci byla opakovaně prokázána přítomnost acetylcholinesterasy jako součást amyloidních fibril.
Následky deposice amyloiduO následcích deposice amyloidu pro biologii tkáně je známo velmi málo. Více méně tradičně se předpokládá interference se zásobením krví a transportem mezi buňkou a kapilárou. Empiricky je znám vliv na propustnost basální membrány glomerulů pro proteiny (masivní proteinurie), který je běžným projevem renální amyloidosy postihující glomerulus). Mechanismus toxického působení amyloidu na tkáň není stále zcela jasný. Amyloidní beta protein Alzheimerovy nemoci, aplikován experimentálně do mozku, je toxický pro neurony mnohem více ve formě fibrilární než ve formě amorfní. Stále větší počet studii však ukazuje, že za toxicitu jsou obecně odpovědny solubilní, oligomerní (prefibrilární) agregáty.
Degradace amyloidu.Amyloidosy jsou progresivní stavy, terapeuticky velmi obtížně ovlivnitelné, s výjimkou odstranění vyvolávající příčiny, pokud je toto možné. Příčina resistence na proteolysu je pravděpodobně mnohočetná. Hlavní příčinou je kompaktnost fibrilární struktury. Nově se však uvádí, že k resistenci na proteolysu významnou měrou přispívá přítomnost vysoce komplexní molekuly SAP (serum amyloid protein P). Vzniklý amyloid neindukuje makrofagickou fagocytární reakci. Lze předpokládat extracelulární proteolysu secernovanými proteasami, která je však významně inhibována přítomností SAP (viz shora). Pro možnost pomalé spontánní degradace svědčí úbytek amyloidu vzniklého na podkladě beta2 mikroglobulinu u hemodialysovaných pacientů při renálním selhání po transplantaci ledvin. To naznačuje existenci určité rovnováhy mezi tvorbou a degradací amyloidu in vivo. Nemožnost indukovat degradaci amyloidu in vivo je limitujícím faktorem terapie amyloidosy. Prioritním požadavkem terapie je proto vyloučení příčiny, možné zejména u klasických sekundárních povšechných amyloidos na basi SAA proteinu. Pokud je příčinou mutace proteinu produkovaného játry byla situace řešena transplantací jater, na př. u familiární ATTR. Pozoruhodné je, že úbytku amyloid beta proteinu v mozkových placích u Alzheimerovy nemoci bylo dosaženo aktivní nebo pasivní imunizací proti tomuto proteinu i přes existenci hemoencefalické bariery a to jak experimentálně na myším modelu, tak i u prvních pacientů. Existuje předpoklad, že protilátka namířená proti solubilnímu prekursoru může působit proti procesu agregace do fibril. Nelze do budoucna vyloučit terapeutické využití Kongo červeně pro její schopnost molekulárně interagovat s amyloidní fibrilou a potencionálně snižovat její stabilitu.
Průkaz amyloidu a jeho specifikace
Diagnostika amyloidu má dva stupně, zcela analogicky jako u onemocnění spojených s deposicí krystalů :
1. Fyzikální průkaz amyloidu - průkaz amyloidní beta fibrily
2. Průkaz biochemické povahy - amyloidogenního proteinu
ad1. Průkaz fyzikální specifity amyloidu se opírá o průkaz fibrily složené z v konformaci beta struktury. Je k tomu využíváno látek, jejichž molekula má k beta doménám fibrilárně agregovaného proteinu afinitu. U nejpoužívanější techniky využívající Kongo červeně se nehodnotí barevný efekt vazby, tak jak je tomu běžně v histochemii. Hodnotí se indukovaný (nebo výrazně zesílený) dvojlom polarisovaného světla (vlákna amyloidu mohou primárně slabý dvojlom vykazovat) a dichroismus.
Tyto efekty jsou měřitelné díky pravidelné adsorpci molekul barviva (nejspíše prostřednictvím vodíkových vazeb) na povrch amyloidních fibril. Asymetrická beta struktura indukuje dva zmíněné optické efekty v polarizovaném světle: dvojlom a dichroismus. Barvivo tento efekt zesiluje a umožňuje jej měřit ve viditelném světle Dichroismus je definován jako schopnost absorbovat část viditelného spektra polarizovaného světla. Dichroická látka se tedy může stát barevnou ve světle polarisovaném, pokud je v běžném nepolarizovaném světle bezbarvá. Dvojlom polarizovaného světla je známou schopností krystalických látek měnit směr výsledného hlavního vektoru vnikajícího polarizovaného světla na směr jiný.
Podle Bubise a Wolmana je fenomen zeleného zbarvení amyloidu Kongo červení závislý do značné míry na síle řezu. Zelený dichroismus je vázán na průměrnou slíu řezu 5-10µ. Řezy slabší (1-2µ) a silné (okolo 20µ) zelený dichroismus amyloidu nevykazují. U tenkých řezů jde tón více do modré, u silných řezů více do žluta, oranžova a u nejsilnějších do červena (negativní dichroismus). Významnou roli hraje rozdílné zpomalení paprsku červeného světla při průchodu řezem (detaily viz Histochemie 4:351-356,1965).
Vazbu Kongo červeně lze velmi citlivě detekovat fluorescenčně, protože molekula barviva sama je dosti silný fluorogen.. Jako nejvhodnější je doporučována sada filtrů pro detekci fluoresceninisothiocyanatu (abs. max. 495; emise max. 525) , méně pro rhodamin (abs. max. 555; emise580). Výsledek je nutno vždy kontrolovat s dichroismem, aby byla vyloučena nespecifická vazba Kongo červeně (t.j. vazba nevedoucí k dichroismu). Metody používající jiné fluorogeny (Thioflavin T a S) jsou považovány za citlivé, ale méně specifické (není kontrola analogická dichroismu).
Fibrilární struktura je výtečně znázornitelná elektronovým mikroskopem. Jde o fibrily nejčastěji 8-10 nm silné, nevětvené, vytvářející různě hustou plsť. Fibrily se skládají z protofilament 2.5 - 3.5 nm silných spirálně se obtáčejících. Jejich počet v jedné fibrile amyloidu může kolísat podle výchozího amyloidogenního proteinu. Šíře fibril tak může kolísat. Maximální šíře byla popsána u TTR (13 nm), minimální u fibril kalcitoninového amyloidu (5-6 nm).
Pro průkaz amyloidu in vivo lze použít scintigrafie s radioaktivně značeným SAP, využívající jeho vysoké afinity k deponovanému amyloidu, jehož je integrální součástí (viz níže). U zdravých kontrol je aplikovaný SAP rychle degradován a degradační produkty vyloučeny močí. Vzácně má amyloidem prostoupený orgán dosti příznačný nález při echografii. Je známý obraz "jiskření" (sparkling) v dvojrozměrném echokardiogramu u amyloidosy srdce (stejný obraz byl popsán u Fabryho nemoci).
Přechodem k detailnější klasifikaci již prokázaného amyloidu bylo empirické pozorování inhibice zbarvení Kongo červení preoxidací řezu permanganátem. Šlo zejména o amyloid na basi AA proteinu (sekundární povšechná amyloidosa, viz níže). Tato sensitivita však byla prokázána i u amyloidu na basi beta2MG.(viz níže) V současné době je tato metoda plně nahrazena imunohistochemickým průkazem.
ad2. Biochemická a imunohistochemická specifikace amyloidu prokazuje proteinový stavební kámen. Dnes v tomto smyslu dominuje imunohistochemie průkazem molekulárně specifického epitopu. Ideální pro detekci je nefixovaná tkáň (kryostatové řezy). Konvenční formaldehydová fixace a zalití do parafinu snižuje možnost imunohistochemické detekce. Za dobře imunohistochemicky prokazatelné v rutinních parafinových řezech se považují amyloid na podkladě SAA a beta2 mikroglobulinu. Detekce AL ve fixovaném materiálu je méně spolehlivá. Dalším důvodem snížené spolehlivosti detekce AL imunohistochemickou cestou je značná přirozená variabilita N terminálních úseků lehkých řetězců (jejich variabilní domény).
Pro imunohistochemický průkaz amyloidů je doporučeno vystavit parafinové řezy koncentrované mravenčí kyselině (nebo alkalickému quanidinu) před vlastní detekcí. Jde o empirický postup, který se osvědčil zejména při imunodetekci cerebrálních amyloidů. Kongofilie je tímto postupem zrušena.
Určité problémy může způsobit výraznější příměs koprecipitovaných komponent, zejména z třídy apoproteinů, které jsou v některých případech známy jako stavební kameny amyloidu, jindy jako koprecipitované komponenty, na př. Apoprotein A1. Není také dostupná metoda, která by určila, zda je daný protein agregovaný do amyloidní fibrily nebo přítomný pouze jako koprecipitující komponenta. Teoreticky lze připustit, že by v amyloidní fibrile mohly být sdruženy fragmenty dvou různých amyloidogenních proteinů. Doposud byla přesvědčivě prokázána kombinovaná amyloidosa z Apo AIV a transthyretinu (viz dále) v odlišných fibrilách.
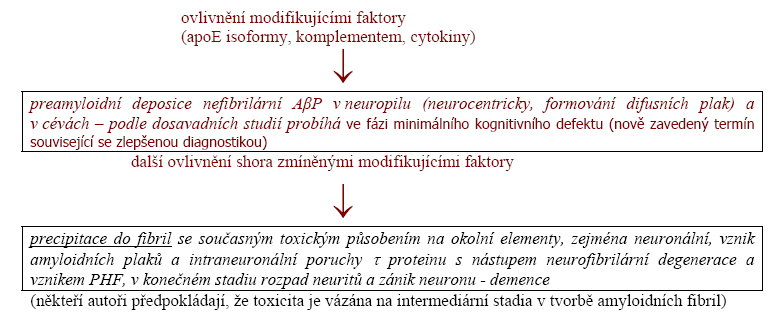
Na tento stav můžeme pohlížet jako na stav preamyloidní (nebo prefibrilární), pokud dojde postupem času ve fibrilární agregaci, nebo jako na stav paramyloidní, t.j. amyloidu histologicky podobnému, ale trvale nefibrilárnímu (historický termín). Výrazná preamyloidní nefibrilární deposice amyloidogenního proteinu je známa u cerebrální amyloidosy na podkladě amyloid beta proteinu a to v t. zv. difusních placích (viz níže). Předpokládá se, že postupem času může pod vlivem řady faktorů do fibril agregovat. V difusních placích se dá detekovat pouze imunohistologicky. Kongo červeň a thiazinová barviva, tedy barviva s molekulární afinitou k beta fibrile dávají negativní výsledek. Stále více prací referuje o přítomnosti nefibrilární formy amyloidogenního proteinu vedle formy fibrilární. Odpovídá to koncepci vzniku amyloidu. Pokud nedochází k fibrilární agregaci chybí asociované komponenty (SAP a další, viz shora).
Amyloid beta protein byl popsán v nefibrilární formě v kosterním svalu myši s vyřazeným apoE genem jako součást membránového systému sarkomerických inklusí ve vláknech typu II. Pravděpodobně zde nedochází k následné konversi na amyloid.
Nejvýraznější nefibrilární deposice amyloidogenního proteinu vzniká v případě imunoglobulinů. Za paramyloidní stav jsou považována jejich amorfní (nefibrilární) deposita, rámcově zvaná onemocnění z deposice monoklonálních imunoglubulinů (MIDD - monoclonal immunoglobulin deposition disease). I u nich byly popsány přechody do amyloidosy. Podrobněji jsou zmíněna níže.
Priony (viz dále) se též akumulují do značné míry v nefibrilární formě v mozcích u prionových onemocnění (viz amyloidosa na basi prionů).
Prionový protein byl též popsán v inklusích (spolu s amyloid beta proteinem) u t.zv. inklusní myositidy (myopatie), patogeneticky nejasného svalového onemocnění starších lidí.
Zvláštní postavení mají t.zv. paired helical filaments vznikající v neuronech CNS z tau proteinu, normálně asociovaného s mikrotululy, tvořící podklad neurofibrilární degenerace neuronů (NFT, neurofibrillary tangles, viz níže). Jejich fysikální vlastnosti jsou identické s amyloidem: barví se Kongo červení, jejíž vazba indukuje zelený dichroismus a jsou nerozpustné. Pouze elektron mikroskopický obraz je od klasických amyloidních fibril odlišný. Jde o zkroucené tubuly, složené z hyperfosforylovaného tau proteinu (viz cerebrální amyloidosy).
Velmi podobné, ne-li strukturálně a fyzikálně identické intracelulární inkluse vznikající v řadě buněčných typů mimo neurony v procesu stárnutí, např. Biondiho tělíska v ependymu a v epitelu plexus chorioideus (viz senilní amyloid).
Kombinovaným použitím imunohistochemického přístupu s barvivy znázorňujícími fyzikální stav se tak lze vyjadřovat ke stupni asemblace amyloidogenního proteinu do fibril.
Amyloidosa není klinicko patologická jednotka - jde o obecný patologický fenomén, vznikající na basi celé řady poruch. Svou obecností a charakterem je srovnatelný s analogickým procesem krystalizace organických či anorganických látek, známým z humánní patologie. Symptomatologie amyloidosy má široké rozmezí od projevů pouze v subklinické úrovni až po rychle progredující fatální stavy. Je určena rychlostí deposice a lokalizací amyloidu, jeho působením na tkáň a případně i příznaky základního onemocnění, v jehož rámci k deposici amyloidu došlo.
Presentace amyloidos je možná podle dvou základních hledisek: podle distribuce (klinicko patologický obraz) a podle amyloidogenního proteinu. Na tomto místě bude kladen větší důraz na amyloidogenní protein a na poruchu, která k jeho fibrilární agregaci vedla. Dalším významným hlediskem je dělení na získané a geneticky podmíněné amyloidosy. Geneticky podmíněné formy jsou charakterizovány dominantním přenosem, pomalou progresí a variabilní penetrancí. Jde o heterozygotní stav s mutací na jedné alele. Druhá alela může nebo nemusí vykazovat mutaci v příslušném proteinu. V případě homozygotního stavu lze předpokládat závažnější průběh amyloidosy.
Čím dále tím více se však ukazuje, že velmi důležitou roli hrají genetické faktory. Jde především o skupinu amyloidos přenosných geneticky, na podkladě mutace řady amyloidogenních proteinů, které mohou mít za následek změněné odbourávání a tím i případně zvýšenou amyloidogenicitu (viz níže). Jde o dominantní způsob přenosu s klinickou manifestaci v dospělosti bez predikovatelného stupně penetrance. Genetická porucha struktury amyloidogenního proteinu může být i příčinou narušeného zpracování v průběhu jeho syntézy a intracelulární fáze životního cyklu (viz např. gelsolin a neuronální amyloid beta protein). Genetické polymorfismy zřejmě hrají roli i v případech SAA a AL reaktivních amyloidos a mohly by vysvětlit, proč dochází ve skupině se stejným základním onemocněním k amyloidose jen u některých pacientů (viz níže). Zvýšená amyloidogenicita se může projevit až za patologických stavů při zvýšené produkci a obratu.
Postavení hemoencefalické bariery. U povšechných viscerálních amyloidos je bariera neporušena a chrání mozkovou tkáň od deposice amyloidu. Jedinou výjimkou mohou být oblasti, ve kterých bariera vytvořena není. Jde o stopku hypofysy a locus coeruleus. Produkce amyloidogenního proteinu na obou stranách bariery vysvětluje současný výskyt amyloidu v mozku i viscerálně, jako je tomu v případě poruchy TTR a cystatinu C dalších (viz níže). V některých případech je postižena takřka selektivně stěna mozkových cév se sekundárním narušením funkce hemoencefalické bariery (viz tzv. kongofilní cerebrální angiopatie). V poslední době se však otevřela otázka transporty fragmentů amyloidogenního proteinu přes hemoencefalickou barieru (viz Alzheimerova nemoc)
K současnému datu je známo spolehlivě 24 amyloidogenních proteinů:
|
Poznámka. V literatuře je započítáván i exogenní injekčně aplikovaný insulin, vytvářející amyloid v místě aplikace.
Tento výčet je s velkou pravděpodobností nekompletní, neboť stále jsou publikována sdělení o amyloidu, který se nepodařilo imunohistochemicky specifikovat.
Na základě rozsáhlých studií lze uzavřít, že beta fibrilární (amyloidní) transformace některých proteinů (typicky TTR, viz níže) probíhá kontinuálně s věkem a to naprosto převážně zcela subklinicky. Nakolik jde o výraz genetického polymorfismu klinicky zdravé populace není známo. Faktem je, že v celé řadě orgánů u lidí staršího věku byly popsány isolované fibrilární agregace různých proteinů (viz níže senilní amyloidosy). V následujícím textu jsou popsány amyloidosy, které jsou projevem klinicky manifestního chorobného stavu.
Zpět na obsahAmyloid na basi sérového AA (amyloid associated) proteinu (SAA). SAA patří do skupiny HDL apolipoproteinů. Jde o reaktant akutní fáze, produkovaný v játrech po stimulaci makrofagickými cytokiny. Byla však prokázána jeho produkce i jinými buněčnými typy. Jeho koncentrace u zánětlivých stavů se může zvýšit během několika hodin řádově až 1000x. Váže se na HDL a zvyšuje jeho vazbu na makrofágy v zánětlivých ložiscích, čímž potencuje významně transport cholesterolu sekvestrovaného v zánětlivých ložiscích do jater.
Jde obecně o proces konstitucionální sekrece hepatocyty (viz kapitolu o sekreci), jejíž intensita je regulována především na transkripční úrovni. U člověka existují 3 transkripčně aktivní geny a jeden pseudogen. Pouze SAA 1 a 2 reagují v akutní fázi zánětu . SAA 3 je nepřepisovaný pseudogen, SAA 4 je trvale produkován jen v basálním množství. Transkripční regulace SAA1 a SAA2 je v dnešní době velmi detailně prostudována a zahrnuje komplexní interakce interleukin-responsivních transkripčních faktorů. Transkript i SAA1 protein byly prokázány i v lidských makrofázích, údajně i v adipocytech a dalších buňkách, ale v podstatně menší míře. Maximum syntesy je v hepatocytech.
Amyloidogení je pouze SAA1, který je však v populaci poměrně polymorfdní. Existují pozorování, podle kterých amyloidogenicitu významně zvyšují některé genetické polymorfismy vedoucí k zvýšené nestabilitě proteinu a jeho precipitaci do fibril.
S vývojem SAA amyloidosy je spojena zvýšená sérová koncentrace SAA. V SAA amyloidu se kumulují neglykosylované N-terminální části o mol. hmotnosti 6-9 kD (v průměru 76 aminokyselin, ale i delší a kratší). Významnou příměs může tvořit i kompletní molekula SAA (mol. hmotnost přibližně 12 kD, obsahuje 104 aminokyselin). V patogenese hraje zřejmě důležitou roli degradabilita SA proteinu v makrofázích, které představují hlavní degradační elementy.
U myšího kmene s vrozenou resistencí na vyvolání experimentální amyloidosy vykazovaly makrofágy vysokou kapacitu degradace SAA, oproti kmenům vnímavým, u kterých byla kapacita degradace podstatně nižší.
Amyloidosa na basi SAA je prototypem sekundární generalizované amyloidosy. Postihuje celou řadu vnitřních orgánů, zejména ledviny, GIT, slezinu, nadledviny s výjimkou CNS. K poruše degradace SA proteinu a k jeho fibrilární agregaci dochází při chronických zánětech, zejména při rheumatoidní arthritidě a některých maligních nádorových procesech. Jde však pouze o malé procento výskytu, naznačující, že ve hře jsou další faktory. Byla popsána i jako forma primární. Doposud však nebyla u člověka popsána primárně geneticky podmíněná varianta. Prototypem experimentálně indukovaná SA amyloidosy je amyloidosa, vyvolaná opakovanou aplikací kaseinu.
SA amyloid však provází některé geneticky podmíněné poruchy, avšak jako sekundární komplikace. Jde o
Amyloid na basi prealbuminu (transthyrethrinu) ATTR (TTR transthyrethrin). Prealbumin je normální komponentou plasmy (název má od postavení na elektroforeogramu plasmatických bílkovin před albuminem). Název transthyretin pochází od toho, že funguje jako přenašeč hormonů štítné žlázy a retinolu (trans - thyr - retin). U zánětů se jeho koncentrace snižuje (nazývá se také negativní protein akutní fáze).
TTR je produktem jediného genu, není glykosylován a vykazuje dosti značný polymorfismus. Normálně je ve formě homotetrameru. Monomer má významně zastoupenou beta strukturu (Obr.1). Asi 90% proteinu je produkováno játry (konstitucionální sekrece na vaskulárním pólu), zbytek v plexus chorioideus (sekrece do mozkomíšního moku) a v sítnici (sekrece do sklivce). Poločas v plasmě je poměrně krátký (1.5 - 2.5 dne). Degradován je z větší části v hepatocytech, méně ve svalové tkáni a v kůži. Vychytávání je na podkladě receptorově zprostředkované endocytosy.
Pro zahájení tvorby amyloidní fibrily je nutná disociace tetrameru na monomery za podmínek mírné denaturace při sníženém pH. Naopak amyloidogenicitu TTR významně v experimentu in vitro snižovala vazba tyroxinu, která stabilizuje tetramer a brání jeho disociaci a následným konformačním změnám.
Vznik amyloidu významně zvyšují mutace v primární struktuře proteinu, podmiňující zvýšenou konformační nestabilitu při denaturaci. V současné době je známo několik desítek mutací, které různou měrou zvyšují amyloidogenní potenciál. Stavy spojené s deposicí TTR lze rozdělit na získané a geneticky podmíněné.
Mezi získané patří t.zv. senilní amyloidosa srdce, zjistitelná u 25% lidí nad 80 let věku, která může vést k mírné formě kardiomyopatie. I za takto mírnou formou se však může skrývat mutace TTR. Amyloidní fibrily se skládají z intaktních molekul TTR ve směsi s menšími peptidovými fragmenty.
Geneticky podmíněné TTR amyloidosy mají autosomálně dominantní přenos. Maximum deposice TTR amyloidu je v periferních nervech a v myokardu, méně v ostatních tkáních. Postižení ledvin bylo popsáno, převážně však v oblasti dřeně. Vzhledem k výraznému postižení nervů je známá pod názvem familiární amyloidní polyneuropatie (FAP). Opakovaně byla popsána klinicky závažná leptomeningeální amyloidosa a deposice amyloidu ve sklivci, pravděpodobně jako následek syntézy TTR epitelem plexus chorioideus a sítnicí. Ke klinické manifestaci dochází v nižším věku. Jsou známy akcelerované průběhy s manifestací ve druhém nebo třetím decenniu. Hladina TTR v plasmě je snížená.
Přídatná (mimojaterní) místa syntézy TTR jsou nejspíše příčinou trvající produkce mutantního TTR u případů léčených transplantací jater.
U geneticky podmíněných TTR amyloidos s mutací v heterozygotním stavu je to směs normálních a mutantních forem proteinu.
Amyloid na basi lehkých řetězců imunoglobulinů (AL amyloidosa, A=amyloid, L= lehké řetězce)Pozn. k fysiologii Ig. Popis synthesy sekrece Ig molekuly je zcela mimo rámec tohoto textu. Zčásti odkazuji na základní charakteristiky tohoto procesu, zmíněné v kapitole sekrece, týkající se problému kompletace molekuly Ig jakožto nezbytného předpokladu pasáže sekretorickou cestou. Pokud jde o degradační procesy v obratu Ig (volného nebo po vazbě na antigen) je detailních studií velmi málo. Lze předpokládat, že degradace Ig probíhá intracelulárně v lysosomálním systému mononukleárních fagocytů, do kterých se dostává receptorově zprostředkovanou endocytosou (přes Fc receptor). O možné extracelulární proteolyse Ig není známo nic. Lehké řetězce Ig (kapa a lambda) patří mezi proteiny velmi bohaté na beta řetězce, což přispívá k jejich potenciální amyloidogenicitě. Základní schema molekuly Ig a jejích amyloidogenních částí je na Obr. 2.
Stavy vedoucí k deposici imunoamyloidu jsou charakterisovány přítomností abnormálního klonu B lymfocytů, produkujícího monoklonální Ig. Jde buď o klon nádorový (nejčastěji jde o mnohotný myelom, nebo obecně o sekrečně aktivní imunocytom) nebo nenádorový (t.zv. primární generalisovaná AL amyloidosa charakteru MGUS - monoclonal gamapathy of undetermined significance).
Biochemická analysa AL amyloidu ukázala přítomnost N-terminálních částí lehkých řetězců (variabilních úseků) nebo jejich fragmentů, vzácně celých lehkých řetězců (tedy variabilní i konstantní části, viz Obr. 2). Ukazuje se, že k amyloidose vede dvakrát častěji typ konfigurace lambda než kappa. Významným predisponujícím faktorem k fibrilární agregaci kritických amyloidogenních fragmentů Ig je také syntéza nekompletní molekuly Ig. V séru (a v moči) pacientů jsou pak prokazatelné samotné monoklonální lehké řetězce (variabilní a konstantní části), známé pod historickým názvem Bence Jonesův protein (BJP) (objevil ho r. 1845 Dr. Henry Bence Jones). Tento abnormální produkt je vysoce risikovým faktorem v dalším rozvoji nemoci. Jeho patogenita závisí na možnosti jeho další agregace (kovalentní přes SS můstky, ale zřejmě i nekovalentní), indukující dimerisaci. Vzájemná afinita lehkých řetězců podmiňující stupeň jejich agregace závisí na charakteru složení variabilních částí. Jednak jde o variabilitu primární, odrážející strukturu antigenního epitopu (podklad idiotypové variability). Touto primární fysiologickou variabilitou se lehké řetězce diametrálně odlišují od molekulárně uniformních jiných amyloidogenních molekul, na př. transthyretinu (viz shora). Dále se ukazuje, že záměny aminokyselin v dalších úsecích (podmíněné zárodečnými nebo somatickými mutacemi) vedou k snadné destabilizaci terciární struktury a k agregaci, na jejímž konci je fibrila amyloidu . Jde o komplementární úseky (CR1-3) a o úseky mezi ně vmezeřené (FR - framework regions). Lze tedy předpokládat i geneticky podmíněnou predisposici ke vzniku imunoamyloidu. Tyto skutečnosti jsou vysvětlením poměrně nízké incidence amyloidosy u mnohočetného myelomu, která je odhadována pouze na 10-15%.
Vysokou asociaci s imunoamyloidem vykazuje Asp50 (v oblasti CDR2), Asp31 (v oblasti CDR1) a obzvláště náhrada prolinu v posici 4O aminokyselinou hydrofobního typu v konservované vmezeřené FR2 oblasti. Jsou určité předpoklady, že existují další posttranslační modifikace zvyšující amyloidogenicitu lehkých řetězců, jako stupeň jejich glykosylace. Není rovněž známo, nakolik jsou lehké řetězce v depositech amyloidu modifikovány proteolysou.
Přítomností cirkulujících přímých prekursorů se patogenese imunoamyloidu odlišuje od předchozích generalisovaných amyloidos, u kterých cirkulují v krvi kompletní normální nebo mutované molekuly prekursorů amyloidu (viz SAA a TTR amyloidosu). Ani u imunoamyloidu však nelze vyloučit možnost vzniku amyloidogenních fragmentů proteolysou kompletní monoklonální molekuly Ig v periferních tkáních.
Patogeneticky významná jedinečnost cirkulujících fragmentů lehkých řetězců Ig u případů s AL amyloidosou byla prokázána experimentálně. Myším byly injikovány monoklonální lehké řetězce isolované z močí pacientů s AL amyloidosou a bez AL amyloidosy. Mnohem výraznější amyloidosa s deposicí převážně intaktních použitých lehkých řetězců byla vyvolána užitím BJP z pacientů s amyloidosou, zatímco v případě moče pacientů bez amyloidosy byly výsledky zcela rudimentární.
Opakovaně byly popsány imunocytomy s monoklonální gamapatií a rozsáhlou intracelulární deposicí fibrilárně uspořádaných lehkých řetězců. Část intracelulárních deposit bylo v endoplasmatickém retikulu a v membranosních systémech náležejících sekretorické cestě, část v makrofázích a v lysosomálním systému celé řady buněčných typů. Interpretace těchto výjimečných nálezů je obtížná (viz níže).
AL amyloidosa je známa jako forma generalizovaná a forma lokální tumoriformní. Distribuce u generalizované formy je povšechná. Agregace do fibril probíhá prakticky ve všech orgánech, s výjimkou CNS. Maximum deposice je v ledvinách, myokardu, slezině, nadledvině, může být v játrech, plicích, ale i v měkkých tkáních (kůži, kloubech, karpálním tunelu). Bývá makroglosie. Může být postižen periferní nervový systém.
Forma lokální (tumoriformní) se vyskytuje v řadě orgánů (na př. hrtan, plíce, gastrointestinální trakt, ale i mozek). Nikdy nelze zcela vyloučit, zda nejde o projev současně probíhající generalisaci amyloidosy
Amyloidogenní fragmenty Ig mohou agregovat alternativním, neamyloidním způsobem. Jedním z nich, velmi běžným, je agregace BJP ve formě hyalinních válců resp. proteinových krystalků v renálních tubulech, vedoucí k t. zv. myelomové nefrose na podkladě nefrohydrosy. Tato agregace předpokládá přesycení resorpčního mechanismu v proximálních tubulech a precipitaci s Tamm-Horsefalovým glykoproteinem v distálním nefronu.
Další alternativní deposicí je amorfní generalizovaná deposice monoklonálních lehkých řetězců Ig (zejména typu kappa) v neamyloidní amorfní formě. Svým výrazně hyalinním vzhledem však amyloid připomíná. Maximum deposice je v basálních membránách. Barvení Kongo červení je však negativní. V elektronovém mikroskopu jsou deposita amorfní, filamentosní (bez beta struktury) nebo mikrotubulární. Od amyloidu je odlišuje, vedle negativity v barvení Konžskou červení, též absence sérového amyloid P proteinu. Pro tato deposita byl v historii použit název paramyloid. Vzhledem k tomu, že ve výjimečných případech byla prokázána deposice celého Ig, tedy i těžkých řetězců je tento stav nazýván "onemocnění z deposice monoklonálních imunoglobulinů" (monoclonal imunoglobulin deposition disease, MIDD). Spojení agregace amorfního typu a typu fibrilárního bylo popsáno u několika pacientů a svědčí o existenci dalších, pravděpodobně lokálních tkáňových faktorů, které se podílejí na typu výsledné agregace. Agregace Ig nastává za těchto stavů generalizované.
V nefropatologii spadá MIDD do širší kategorie fibrilárních/mikrotubulárních glomerulopatií (Kongo negativních), které mohou být součástí generalizovaného postižení. Tyto procesy jsou nepochybně heterogenní co do patogenese a rozlišování je stále závislé na morfologii deposit, na kterou zde nelze zacházet (viz literatura).
Poslední známou variantou osudu monoklonálního Ig je stav spojený s jeho excesivní a prakticky kompletní endocytosou makrofágy (zejména v okolí nádorových ložisek). Proto mezinárodně užívaný název "crystal storing histiocytosis" (i když o krystaly vždy nejde). Velmi často se na endocytose podílí celá řada dalších buněk, takže jde pak o generalizovaný proces. Takovýto stav lze popisně nazvat generalizované lysosomální střádání monoklonálního Ig. V lysosomech endocytovaný protein v krystalické, amorfní nebo fibrilární formě. Fibrily jsou odlišné of fibril amyloidních.
Za toho stavu dochází také k toxickému poškození tubulárních funkcí ledvin endocytovaným monoklonálním imunoglobulinem, zejména v oblasti proximálních tubulů. Může dojít až k nekrose.
Imunoamyloid na basi fragmentů těžkých řetězců Ig (AH - amyloid heavy chain) jako kontrast k AL - amyloid light chain). Doposud byly popsány zcela ojedinělé případy. U jednoho z nich šlo o těžkou renální amyloidosu u starší pacientky s deposicí fragmentu těžkého řetězce IgG v mesangiu glomerulů.
Amyloidosa na basi beta2 mikroglobulinu (Abeta2MG). beta2MG je ubikviterní protein lokalizovaný na buněčném povrchu všech buněk v těsné funkční s molekulami MHC I komplexu. Nejde o transmembránový protein. Patří do kategorie malých proteinů (m.v. okolo 12 kDa). Má výrazně zastoupenou beta strukturu. Má konstantní hladinu v extracelulární tekutině. Za normálních okolností je vylučován ledvinami. Amyloidní agregace beta2MG nastává za stavů trvale zvýšené sérové koncentrace, což nastává prakticky pouze u dlouhodobě dialýzovaných pacientů. Příčinou je skutečnost že neprochází póry hemodialyzační membrány a následně se hromadí v extracelulární tekutině. Tím vznikají, vzhledem k významné přítomnosti beta konformace, předpoklady pro vznik amyloidu. Podle dostupných studií jde asi o 10%-30% výskyt u dlouhodobě hemodialyzovaných pacientů.
beta2MG amyloidosa má poměrně typická predilekční místa deposice. Jde především o synovii, chrupavky (kloubní a meziobratlových destiček) a kostní tkáň. To vede ke klinickému obrazu spondylarthropatie, syndromu karpálního tunelu, často i osteolytických ložisek (nodulární kostní deposita amyloidu). Predisponujícím faktorem je zřejmě přítomnost chondroitin sulfátu, který v experimentu in vitro významně katalysoval fibrilární agregaci beta2MG. Viscerální lokalizace může být rozsáhlá, ale většinou je subklinická. Postiženy jsou především cévy. Na deposici beta2MG amyloidu je pozoruhodně resistentní slezina. Deposita amyloidu obsahují intaktní beta2MG (jako dimer), ale popsány byly i zčásti degradované formy. Tinkčně a ultrastrukturálně jsou deposita typická. Amyloid je citlivý na kalium permanganátovou preoxidaci.
Amyloidosa na basi lysozymu (ALys). Doposud bylo popsáno jen několik případů. Jde o generalizovanou amyloidosu, podmíněnou geneticky (autosomálně dominantní přenos). Bývají masivně postižená zejména játra. Mutace v enzymovém proteinu (Ile56Thr nebo Asp67His) způsobuje jeho destabilizaci a fibrilární agregaci. Výsledky analysy složení amyloidu u heterozygotů ukázaly, že se do fibril agreguje pouze intaktní mutovaný protein (nikoliv protein normální).
Amyloidosa na basi alfa řetězce fibrinogenu (AFib). Alfa řetězec je největší ze všech tří řetězců fibrinogenu. Je tvořen 610 aminokyselinami. Amyloidosa na basi tohoto proteinu je známa pouze v dědičné formě. Přenos je autosomálně dominantní. Postihuje celou řadu vnitřních orgánů (játra, slezinu, nadledviny), zejména však ledviny (je také zvána hereditární renální amyloidosa), kde masivně postihuje glomeruly. Periferní nervový systém není postižen. V depositech amyloidu u heterozygotní mutace byla prokazatelná pouze mutantní forma proteinu.
Amyloidosa na basi cystatinu C (ACys). Cystatin C je inhibitorem cysteinových proteás (kathepsinů B,H.L). Jde o neglykosylovaný polypeptid o 120 aminokyselinách. Je produkován celou řadou buněčných typů, včetně monocytů. Je přítomen ve všech tělních tekutinách, včetně cerebrospinálního likvoru (koncentrace 6.5 mg/l). Mutantní cystatin C je enormně náchylný k dimerisaci, která je ireversibilní, vede k afunkčnosti proteinu a usnadňuje fibrilární agregaci. Amyloid obsahující degradační produkty mutantního proteinu se deponuje generalizovaně. Jde o systémovou amyloidosu s postižením řady orgánů i mimo oblast jejich cévní sítě. Po diagnostiku je využívána kůže, ve které je deposice prokazatelná v oblasti basálních membrán okolo kožních adnex, okolo adipocytů a ve stěně cév. Masivně jsou však postiženy meningeální a cerebrální cévy, což je podkladem cévních ruptur a mozkových hemorrhagií. Jednotka byla nazývána hereditární cerebrální amyloidová angiopatie - islandský typ vzhledem k topice příznaků. Výstižnější je biologický termín systémová amyloidosa na basi cystatinu C. Příznačná je snížená koncentrace cystatinu C v tělních tekutinách. V neuropilu k deposici amyloidu nedochází (může být však přítomna nespecifická deposice amyloid beta proteinu, úměrná věku).
Monocyty pacientů vykazovaly v tkáňové kultuře sníženou sekreci cystatinu C, což by mohlo vysvětlit jeho sníženou hladinu v extracelulární tekutině
Cystatin C se však může sekundárně akumulovat v depositech amyloid beta proteinu u Alzheimerovy nemoci.
Amyloidosa na basi ApoAI (AApoAI). ApoAI je kvantitativně nejvíce zastoupený apoprotein třídy HDL. Jde o protein o 243 aminokyselinách, produkovaný enterocyty, hepatocyty a celou řadou dalších buněk. Mutantní protein je poměrně vysoce amyloidogenní. Deposice amyloidu je generalizovaná, s maximem v ledvinách (časté je renální selhání), játrech a srdci (bývá fatální kardiomyopatie) a ve slezině. V ledvinách bývá maximum deposice mimo glomeruly. Velmi často se popisuje kožní deposice amyloidu (difusní nebo papulární). Též postižení laryngu bylo opakovaně popsáno. Běžně se nazývá non-neuronopatická dědičná generalizovaná amyloidosa, i když postižení periferních nervů bylo opakovaně popsáno. Postižení se manifestuje již v heterozygotním stavu (autosomálně dominantní přenos) a může se manifestovat v mládí již od druhého decennia). V amyloidu byl ve všech doposud studovaných případech přítomen N terminální úsek mutantního proteinu, obsahující přibližně 80 aminokyselin a to i v amyloidních depositech u heterozygotů (divoký protein, produkt divoké alely, na amyloidose neparticipoval). U pacientů s renálním a kardiálním selháním se osvědčila transplantace.
Amyloidosa na basi ApoAI (AApoAI). ApoAI je kvantitativně nejvíce zastoupený apoprotein třídy HDL. Jde o protein o 243 aminokyselinách, produkovaný enterocyty, hepatocyty a celou řadou dalších buněk. Mutantní protein je poměrně vysoce amyloidogenní. Deposice amyloidu je generalizovaná, s maximem v ledvinách (časté je renální selhání), játrech a srdci (bývá fatální kardiomyopatie) a ve slezině. V ledvinách bývá maximum deposice mimo glomeruly. Velmi často se popisuje kožní deposice amyloidu (difusní nebo papulární). Též postižení laryngu bylo opakovaně popsáno. Běžně se nazývá non-neuronopatická dědičná generalizovaná amyloidosa, i když postižení periferních nervů bylo opakovaně popsáno. Postižení se manifestuje již v heterozygotním stavu (autosomálně dominantní přenos) a může se manifestovat v mládí již od druhého decennia). V amyloidu byl ve všech doposud studovaných případech přítomen N terminální úsek mutantního proteinu, obsahující přibližně 80 aminokyselin a to i v amyloidních depositech u heterozygotů (divoký protein, produkt divoké alely, na amyloidose neparticipoval). U pacientů s renálním a kardiálním selháním se osvědčila transplantace.
Divoký protein se deponuje v aortální intimě a je spolu s medinovou amyloidosou v medii aorty (viz amyloidosa na basi medinu) podkladem aortální amyloidosy závislé na věku. Amyloid se deponuje zčásti ve formě difusní, zčásti ve formě mikronodulární. Existují náznaky, že by ApoA1 intimální amyloid mohl vykazovat vazbu na stupeň aterosklerosy. Deposice amyloidu na basi ApoA1 (N terminální části proteinu) byla prokázána i meniscích kolenního kloubu jako isolovaný proces. Amyloidní agregace divokého proteinu ApoA1 je podkladem plicní amyloidosy psů.
Amyloidosa na basi Apo AII (AApoAII). Jde o geneticky podmíněnou amyloidosu, postihující ledviny a myokard. Zdůrazňuje se postižení cévní stěny v mnoha orgánech. Doposud byly popsány ojedinělé případy.
Amyloidosa na basi Apo AIV (AApoAIV) nebyla zatím popsána v samostatné formě. Amyloid na teto basi byl nedávno popsán jako součást kombinované amyloidosy (kodeposice u ATTR) u velmi staré ženy. Oba amyloidy se deponovaly nezávisle. Nešlo o mutantní proteiny.
Amyloid na basi gelsolinu (AGel). Gelsolin je protein regulující růst filamentosního aktinu (aktinu F). Obecně je považován za jednoho z regulátorů motility buněk. Defekt je provázen sníženou buněčnou motilitou. Hlavním místem jeho působnosti je tedy cytosol. Vedle toho však existuje sekreční forma gelsolinu, jejíž funkcí v plasmě je ochrana před aktinem uvolňovaným z rozpadlých buněk, který by spontánně excesivně polymerizoval. Obě tyto formy jsou alternativním produktem jediného genu. Jedna z forem si uchovává signální peptid a přechází do sekreční dráhy buňky (sekreční, sérová forma), druhá zůstává v cytosolu. Normální koncentrace sérového gelsolinu je okolo 220 ľgr/lm. Cytoplasmatický gelsolin je produkován celou řadou buněčných typů, zejména buňkami svalové tkáně.
Amyloidosa na basi gelsolinu je autosomálně dominantní onemocnění s rohovkovou dystrofií a polyneuropatií, počínající většinou jako paresa horní větve lícního nervu. Amyloid se za těchto okolností ukládá v rohovce, v nervech a v některých dalších orgánech. Převládá deposice v basálních membránách cév. Vzhledem k autosomálně dominantnímu přenosu je i heterozygotní stav klinicky manifestní. U homozygotů bývá postižení závažnější. Tato forma amyloidosy patří k prozatím nejlépe probádaným na molekulární úrovni. U gelsolinové amyloidosy (finská familiární amyloidosa) je hlavním patogenetickým momentem mutace v oblasti proteinu, která je společná oběma jeho uvedeným variantám. Sekreční varianta je však v průběhu sekrečního procesu díky mutaci, odkrývající nová štěpná místa, odlišně proteolyticky zpracována. Tím je vyštěpen právě amyloidogenní úsek 71 aminokyselin, který agreguje do amyloidních fibril. Tímto abnormálním proteolytickým zpracováním musí vzniknout i deficit protektivní funkce sérového gelsolinu. O případné dysfunkci mutantní cytoplasmatické isoformy nejsou přesvědčivé doklady.
Gelsolin byl imunohistologicky prokázán v neuronálních Lewyho tělíscích.
Zpět na obsahJde o stavy charakterizované deposicí amyloidu, jehož stavebním kamenem je transmembránový glykoprotein (ev. jeho sekreční forma), endokrinně aktivní polypeptid nebo průvodní peptid sekrečního granula. Společným rysem je lokální charakter amyloidu. Fibrily amyloidu jsou deponovány v pericelulárním prostoru buňky produkující amyloid. Za významný podpůrný faktor se považuje nadměrná stimulace k sekreci amyloidogenního proteinu, případně jeho více amyloidogenní molekulární varianty a přítomnost látek podporujících fibrilární agregaci. Určitou výjimkou v této skupině jsou prionové amyloidy, které jsou však výrazně topicky omezené na CNS. Patří se zdůraznit, že i amyloidosy projevující se standardně generalizovaně mohou mít lokální isolovanou variantu, jak to bylo opakovaně popsáno u AL amyloidosy. Vysvětlení proto chybí, nutno však předpokládat, že monoklonální Ig je efektivně agregován do amyloidních fibril v okolí patologického klonu B lymfocytů.
Cerebrální amyloidosy (neurocentrické a angiocentrické) na basi t. zv. cerebrálního amyloidního prekursorového proteinu (APP). Tyto amyloidosy jsou topicky omezené na mozkovou tkáň, přesto, že kritický amyloidogenní protein je ubikviterní.
APP je transmembránový protein, produkt jediného genu (lokus je na 21. chromosomu). Jeho struktura odpovídá v zásadě membránovému receptoru. Skládá se se z 695-770 aminokyselin. Variabilita je dána sestřihovými variantami, s vystřižením exonů 7, 8 a 15 v různé kombinaci (obr. 4).
Terminologie: APP amyloid prekursor protein, APP695/751/770 isoformy APP s uvedenou délkou aminokyselinového řetězce, sAPP sekretorická forma APP proteinu, AbetaP amyloid beta protein (též betaA4 kde číslovka odráží mol. hmotnost 40 kD), AbetaP1-43 informace o maximální délce řetězce. Čísla udávají počet aminokyselin (zde maximální), P3 je fragment fysiologické cesty štěpení alfa sekretasou (jde o fragment AbetaP17-42)
APP695 (vystřiženy exony 7 a 8) je považován za typickou neuronální isoformu.
APP751/770 isoformy jsou převážně koncentrované v nonneuronálních elementech. Jde o celou řadu tkání, na př. tkáň svalovou, aortu, pankreas, leukocyty, zvláště pak alfa granula destiček (viz níže nexin). V mozku jsou tyto isoformy zejména silně zastoupeny v glii. Jejich sekretorická forma APP (viz níže) má neuroprotektivní a obecně neurotropní účinek.
N konec APP a převážná část řetězce je extracelulární, na C konci je krátký transmembránový úsek zakotvený do buněčné membrány. Do cytoplasmy ční jen nepatrný terminální C úsek. Protein má několik funkčních domén. Kritickým amyloidogenním úsekem je pericelulární úsek, tvořený 39-43 aminokyselinami, zčásti zakotvený do buněčné membrány, nazývaný přímo "amyloid beta protein" (AbetaP).
V písemnictví je zmiňováno několik dalších variant AbetaP. T. zv. appican je sestřihová varianta, s navázaným řetězcem chondroitin sulfátu (sestřihová varianta s vystřiženým 15. exonem). Takto vzniklý variantní produkt APP představuje proteoglykan. Je ubikviterní, v mozku však byla jeho produkce prokázána pouze v astrocytech.
Nexin II je varianta APP obsahující jednu z variabilních domén APP, t. zv. Kunitzův proteasový inhibitor, KPI). Jde o sekretorickou variantu výše uvedených isoforem APP751/770. Má charakter proteasy, přítomné v celé řadě tkání, včetně mozku. Má důležitou roli v hemokoagulaci. Je bohatě zastoupen v alfa granulích krevních destiček.
Funkce APP. V neuronech hraje transmembránový APP pravděpodobně roli v synaptogenesi v průběhu embryonálního vývoje a má nepochybně řadu funkcí, spojených s přenosem vzruchu a protekcí neuronů před inzulty nejrůznějšího druhu. Sekretorická forma je indukovaná depolarizací neuronální membrány a má zřejmě roli v přenosu vzruchu. Někteří se domnívají, že AbetaP moduluje, po uvolnění do extracelulárního prostoru, aktivitu receptorů. Nic není známo o funkci isoforem APP v glii, ve které je bohatě exprimován, zejména ve formě asociované s chondroitin sulfátem (appican).
Velmi propagovaná je teorie, podle které hraje transmembránový APP roli v mezibuněčném kontaktu neuronálních i non-neuronálních buněk. Funkce APP v jiných buněčných typech je známa velmi málo s výjimkou nexinu II (viz shora).
APP byl prokázán v sekrečních granulích neuroendokrinních buněk bovinní dřeně nadledvin, tedy v buněčném sekretorickém systému velmi blízkém sekreci neuronální (viz oddíl sekrece), ve které hraje APP zřejmě velmi důležitou roli (viz níže).
Syntesa APP probíhá klasickým způsobem na ribosomech, pomocí signálního peptidu je translokován do endoplasmatického retikula, dále do Golgiho aparátu. V transgolgi zoně se APP transportuje jako transmembránový protein konstitucionální sekrecí na buněčnou membránu, do které je inkorporován. V neuronech je transportován axonálním transportem do synaptických oblastí nejen intracerebrálně, ale i do periferních nervů. Narušení axonálního transportu může mít za následek stagnaci APP v průběhu axonu. V presynaptických oblastech je lokalisován na axolemě a participuje na recyklaci synaptických váčků spolu s jejich integrálními membránovými proteiny (synaptofysinem, synaptogaminem a SV2), od kterých se má následně oddělit a putovat cetrifugálně do oblasti perikarya neuronu. Do dendritů je transportován velmi pomalu. U sekretorické varianty podléhá parciální proteolyse sekretasami již v průběhu sekreční cesty (viz dále).
Vedle transmembránové formy APP existuje fysiologická forma sekreční (sAPP), vznikající buď odštěpením prakticky celé pericelulární části transmembránové (in situ) lokalizovaného APP nebo již v průběhu sekretorické cesty. Tato proteolytická úprava se odehrává buď in situ v buněčné membráně (údajně však probíhá převážně v nonneuronálních elementech) nebo k ní může dojít, jak se ukazuje, již v průběhu sekreční cesty, tedy intracelulárně (zejména v neuronech, viz níže). Veškerá známá proteolytická úprava APP je realizovaná třemi proteasami, t. zv. sekretasami (alfa, beta, gama). Sekretasy alfa a beta odštěpují extracelulární část APP od části zakotvené transmembránově, čímž vedou ke vzniku sekretorických forem. Alfa sekretasa štěpí APP v oblasti 16. aminokyseliny AbetaP segmentu - zhruba uprostřed kritického amyloidogenního úseku. Její aktivita tedy velmi dobrou prevencí vzniku celistvého amyloidogenního AbetaP. Variantní "processing" beta sekretasou, zejména v kombinaci s gama sekretasou vyštěpí kritický amyloidogenní AbetaP segment (viz obr.13). Poměr mezi fysiologickou alfa úpravou a ostatními variantními amyloidogenními úpravami se může za různých okolností měnit. Je známo, že AbetaP je konstitucionálně produkován i za normálního stavu a v malém množství prokazatelný v extracelulární tekutině, včetně mozkomíšního moku. Jeho produkce může kolísat v závislosti na některých stresových buněčných situacích (viz níže).
K fixaci amyloidogenní cesty se zvýšenou produkcí kritického AbetaP dochází autosomálně dominantními mutacemi v AbetaP proteinu (zejména v oblasti proteinu obsahujícího štěpné místo pro alfa sekretasou) a v presenilinech - proteinech asociovaných doposud ne zcela jasným způsobem s úpravou, transportem a cílením APP buňkou (viz níže).
Uspokojivé vysvětlení amyloidogenní cesty na úrovni buňky však doposud chybí.
Existují pozorování, dle kterých je amyloidogenní AbetaP (zvláště pak delší varianta o 42 aminokyselinách) uvolněn z APP již v průběhu translokace endoplasmatickým retikulem, či Golgiho aparátem, tedy před insercí APP do buněčné membrány a nezávisle na lysosomálním systému. Současně to ukazuje, že příslušné proteolytické enzymy mohou APP upravovat již průběhu sekreční dráhy. Je pravděpodobné, že toto předčasné uvolnění AbetaP může být za některých okolností vystupňováno a vést k zvýšené sekreci amyloidogenního peptidu.. Za zmínku stojí i transgenní experimenty prokazující, že změna poměru sestřihových isoforem APP ve prospěch APP770/751 ve smyslu jejich zvýšení oproti normálnímu neuronálnímu APP695 vedly k těžké formě AD. Změna poměru byla vyvolána cílenou mutagenesou v intronech.
Méně jasná je role lysosomálního aparátu v produkci AbetaP amyloidu. Pokud by nedošlo k úpravě transmembránově situovaného APP alfa sekretasou, nastoupila by sekretasa beta, která by oddělila objemný N terminální konec před AbetaP úsekem. Ten by se pak s celým C - terminálním zbytkem molekuly spolu se zakotvující částí buněčné membrány procesem endocytosy dostal do lysosomu, kde by ho uvolnila sekretasa. Některé experimenty však svědčí pro přímé cílení signifikantní části APP do lysosomů již v průběhu sekreční cesty. Zákonitosti těchto procesů jsou však dosud velmi málo známé.
Fragmenty APP byly prokázané v neurolysosomech u některých lysosomálních enzymopatií spojených s tvorbou residuálních lipopigmentových tělísek (neuronální ceroidlipofuscinosy, normální neurolipofuscin - age pigment). Experimentální práce nanačují velmi obtížnou degradabilitu AbetaP v lysosomech, což může k fenoménu lysosomální akumulace amyloidního peptidu přispívat.
V případě cerebrální AbetaP amyloidosy (Abeta) jde obecně biologicky o spontánní proces, probíhající trvale v malé intensitě v dospělosti a vedoucí ve stáří, zcela subklinicky, k amyloidním depositům v t. zv. senilních placích a v cévách. Situace je zcela analogická progresivní deposici amyloidu v řadě jiných orgánů (viz níže). Tento basální spontánní proces může být akcelerován celou řadou faktorů a vést tak k progresivní destrukci neuronální a cévní sítě a tím k závažným klinickým změnám, jejichž dominantním projevem je demence. Tato klinicko patologická jednotka se nazývá Alzheimerova nemoc (AD).
Alzheimerova nemoc ve formě sporadické je nejčastější příčinou demence u starých lidí. Jen asi 2-7% je podmíněno dědičnou poruchou, má familiární charakter a rychlý průběh s manifestací ve středním věku.
Odlišení subklinicky probíhajících senilních změn od subklinické formy AD na základě neuropatologických změn je nemožné, neboť veškeré degenerativní změny (viz níže) jsou společné, takže rozlišení je v současné době možné na podkladě kvantitativních rozdílů (další podrobnosti viz neuropatologické monografie).
Akcelerující faktory nepodmíněné geneticky. Ke zvýšené produkci AbetaP dochází v mozku obecně za stavů označovaných souhrnně jako buněčný stres. Jde o ischémii, stavy spojené s traumatem mozku, či energetickou deprivaci nejrůznějšího druhu, nebo o variabilní lokální faktory typu změny pH, vlivu iontů těžkých kovů, při kterých dochází k aktivaci štěpení beta sekretasou. Pravděpodobně nejdůležitější jsou vlivy, které usnadňují a katalysují fibrilární agregaci nadměrně produkovaného AbetaP. Kumulace těchto faktorů je pravděpodobně podkladem procesu amyloidní deposice spontánně probíhající s věkem. Významným akcelerujícím faktorem je přítomnost isoforem apoE4 (viz níže). Je dobré zdůraznit, že výrazná akumulace nefibrilárního AbetaP byla opakovaně prokázána u velmi starých osob, u kterých nebyly za života patrné žádné známky demence.
Faktory geneticky podmíněné. Přenos je ve všech těchto případech autosomálně dominantní. Jde jednak o významné faktory modifikující a o faktory kausální, podmiňující familiární výskyt, časný vznik a fatální průběh cerebrální amyloidosy (familiární AD - FAD). K současnému datu je akceptováno 11 odlišných genetických faktorů (viz OMIM), z nichž jsou nejprobádanější a nejvýznamnější následující.
Mutace zmíněných proteinů zodpovídají za 70-80% AD s časným začátkem. Průběh je velmi rychlý a fatální. Mechanismus akcelerace nemoci mutacemi v těchto proteinech není doposud objasněn.
Preseniliny jsou transmembránové ubikviterní proteiny o doposud ne zcela známé funkci. Jsou normálními konstituenty neuronálního perikarya. Presenilin 2 je zčásti homologní s presenilinem 1. Současné poznatky svědčí pro jejich úzký vztah ke sekretase v tom smyslu, že oba představují samotný enzym, štěpící intramembránové úseky transmembránových proteinů. Tím je v současnosti nahrazen původní předpoklad jejich neenzymového charakteru. Lze tedy dedukovat, že mutace v presenilinech, vedoucí k urychlené Alzheimerově nemoci, nevedou ke ztrátě katalytické aktivity, ale k převážnému vyštepování amyloidogenního úseku v membráně neuronu. Jde de facto o kvalitativní poruchu sekrečního mechanismu na basi "ectodomain shedding" (viz úvod).
Společným jmenovatelem všech stavů je v prvé řadě zvýšená produkce AbetaP, zejména řetězce o 42 aminokyselinách (AbetaP1-42,.), vystupňovaná zejména u geneticky podmíněných forem nemoci a zejména existence faktorů, usnadňujících jeho fibrilární agregaci.
Agregace AbetaP do fibril je děj poměrně složitý, vyznačující se některými specifickými rysy. Jde především o chemické reakce, při nichž dochází k produkci volných radikálů a k poškození okolních buněk. Tyto průvodní chemické reakce jsou nejspíše jedním z faktorů podmiňujících toxicitu agregujícího (nikoliv již agregovaného) AbetaP. Pro precipitaci amyloidu a vznik mikroskopicky detekovatelných lesí je nutné vytvoření primárního fokusu (nidus) překonáním kritické koncentrace AbetaP, který pak působí jako nukleační faktor. Fibrilární agregace je tedy pouze jedním ze dvou fyzikálních stavů AbetaP, amorfního a fibrilárního. Toto je situace v patofysiologii amyloidu neobvyklá. Naznačuje, že fibrilární agregace vyžaduje v mozkové tkáni speciální podmínky (viz též priony). Tato situace může být v patogenese amyloidos (i mimo Alzheimerovu nemoc ) velmi častá. O positivním vlivu imunizace proti AbetaP na průběh Alzheimerovy nemoci je stručná zmínka v úvodu.
V extracelulární tekutině je AbetaP přítomen řádově v nízkých (nanomolárních) koncentracích a to jak za normy, tak u Alzheimerovy nemoci. V pokusech in vitro však nastává spontánní agregace ABP v koncentracích tisícinásobných. To ukazuje na nutnot existence endogenních faktorů snižujících kritickou koncentraci. Za takovéto faktory jsou považovány některé z molekul, které jsou v depositech prokazatelné.
Pokud jde o samotný AbetaP, varianta o 40 aminokyselinách agreguje do fibril podstatně pomaleji než varianta s 42 aminokyselinami.
Příměsi jsou zčásti stejné jako v amyloidech obecně: sérový protein P, heparan sulfát, produkty astrocytů apoprotein E (zejména zmíněná isoforma 4), působící ve smyslu amyloidogenním, dále apo J (clusterin), opět produkt astrocytů, působící ve směru antiamyloidogenním. Dlouho známou příměsí je C1q komplementu (produkovaný aktivovanou mikroglií) a acetylcholinesterasa.
Lokalizace amyloidu. Je pozoruhodné, že cerebrální amyloidosy, jakožto topicky omezené amyloidosy, mají částečně i rysy generalizované poruchy. Zvýšená produkce AbetaP (nikoliv fibrilární deposice) u geneticky podmíněných forem AD byla prokázána i v kultivovaných kožních fibroblastech. Tato zvýšená produkce je příčinou zvýšené koncentrace AbetaP v tkáňových tekutinách, včetně mozkomíšního moku za těchto stavů.
Experimentálně byla prokázána možnost transportu AbetaP přes hemoencefalickou barieru jak samostatně, tak však zejména v asociaci s apoproteinem J (clusterinem). Rovněž asociován s apoE4 může být transportován přes tuto barieru do CNS (nikoliv v asocioaci s apo E2, E3). Existuje dokonce názor, že AbetaP je produkován extracerebrálně, je transportován do CNS přes hemoencefalickou barieru a v mozku precipitován do fibril.
Deposita AbetaP byla v serii 11 případů nalezena v kůži a ve střevní stěně okolo cév a ve vazivu. Tato deposita měla charakter preamyloidu, t.j. vykazovala specifickou reakci s protilátkou proti AbetaP, ale nebyla kongofilní a tudíž ani fibrilární.
Deposice AbetaP amyloidu v mozku probíhá ve dvou odlišných lokalizacích: ve vztahu k neuronální síti (neurocentricky) a v cévní stěně (angiocentricky). Je to zřejmě výrazem toho, že vedle neuronů, které jsou nepochybně významnými producenty AbetaP je AbetaP produkován i elementy cévní stěny mozkových a leptomeningeálních cév. Současně dochází, na rozdíl od jiných typů cerebrálních amyloidos (viz priony) intraneuronálně k fibrilární transformaci tau proteinů do fibril.
1. Neuropil šedé hmoty - neurocentrická deposice. Hlavním producentem AbetaP v této lokalisaci jsou neurony. AbetaP se hromadí ve dvou odlišných formách.
Jednou z nich je forma amorfní, identifikovaná nedávno, ve které se ukládá buď difusně v určitých oblastech kůry nebo ve formě klasických drobných dispersních ložisek, t.zv. plaků (t.zv. difusní plaky). Tato ložiska nejsou detegovatelná Kongo červení ani Thioflavinem S, pouze imunohistologicky. Je všeobecná tendence je považovat za prekursory plaků amyloidních.
V senilních placích jsou obecně velmi častá větší deposita APP v dystrofických neuritech jako následek narušeného axonálního transportu v degenerujících neuronech (viz shora). V některých neuronech bylo u Alzheimerovy nemoci prokázáno hromadění APP (tedy prekursorového beta proteinu) v neuronálním perikaryu, zejména v nc. basalis Meynerti.
Druhou je forma fibrilární s klasickými amyloidními fibrilami, vykazujícími Kongofilii se zeleným dichroismem a typickou ultrastrukturou v EM. Tato forma je přítomna ve formě plaků, zvaných klasické. Plaky difusní (zvané též některými autory preamyloidní) a klasické (amyloidní) se vyskytují převážně odděleně, jejich překryv je jen částečný. Další detaily o jejich složení a vztahu k dystrofickým neuritům a neurofibrilární degeneraci neuronů přesahují rámec tohoto textu. Ve spektru variant AbetaP v amyloidu neuropilu dominuje AbetaP1-42.
2. Angiocentrická deposice (cerebrální amyloidní angiopatie - CAA, též kongofilní angiopatie). Za hlavního producenta AbetaP ve stěně mozkových a leptomeningeálních cév jsou považovány buňky hladkého svalu a pericyty. U obou byla prokázána schopnost produkce APP. V amyloidních depositech cévní stěny dominují kratší formy AbetaP (AbetaP 40). Amyloid je ukládán primárně do basálních membrán. Deposice amyloidu začíná v zevní části stěny. Postihuje především leptomeningeální cévy a cévy kůry mozkové. Hlouběji situované cévy v mozkovém tkáni jsou postiženy vzácně. Deposice amyloidu je provázena degenerativními změnami, oslabením cévní stěny a poruchou hematoencefalické bariery. Jedním z nejzávažnějších projevů cerebrální amyloidní angiopatie je mozková hemorrhagie, většinou atypicky lokalizovaná (mimo basální ganglia). Deposice amyloidu jde společně s deposicí amyloidu v neuropilu a je tedy integrální součástí Alzheimerovy nemoci. CAA může v některých případech výrazně dominovat (kongofilní angiopatie bez demence je prokazatelná u 60% starších lidí).
V rámci AD existuje varianta zvaná hereditární mozková hemorrhagie s amyloidosou - Dánský typ, představující ustálený fenotyp s naprostou převahou angiocentrických změn nad změnami neurocentrickými. Zodpovědné jsou mutace postihující 21. (glycin za alanin) nebo 22. (glutamová kyselina za glutamin) aminokyselinu AbetaP. Selektivní postižení cerebrálních a leptomeningeálních cév postrádá vysvětlení. V cévách mimo mozek není amyloid na basi AbetaP prokazatelný.
3. Intraneuronální fibrilogenesa. Deposice AbetaP amyloidu je provázena amyloidní transformací neuronálního tau proteinu (protein asociovaný s miktotubuly). Příčinou této transformace je trvale udržovaný hyperfosforylovaný stav tau proteinu (pravděpodobně nedostatečná defosforylace). V tomto stavu nemůže nastat jeho fysiologická asociace s mikrotubuly. Takto hyperfosforylovaný tau protein se následně agreguje do fibril, které vytvářejí intraneuronálně t.zv. párová helikální filamenta (paired helical filaments, PHF), jejichž dalším stadiem jsou NFT (neurofibrillary tangles), ve kterých jsou přítomny vedle PHF i jednoduchá filamenta. Tinkcí odpovídají PHF a NFT amyloidu - jsou kongofilní a indukovaný dvojlom má zelený dichroismus. Odpovídá to průkazu beta struktury roentgenovou difrakcí.
V PHF je vedle tau proteinu přítomen dle některých autorů i AbetaP, alfa1 antichymotrypsin, heparan sulfát, fibroblastový růstový faktor, apoE a ubikvitin. Ubikvitinace naznačuje tendenci k degradaci PHF v proteasomu. Popsána byla i neenzymatická glykosylace proteinu v PHF. Heparan sulfátu se přisuzuje významná role v katalyse ireversibilní fosforylace tau proteinu a tím nepřímo v katalyse dalších změn. Proti extraneuronálnímu amyloidu však údajně chybí SAP.
Struktura analogická NFT byla prokázána v některých nonneuronálních buňkách v průběhu procesu stárnutí (viz senilní amyloid).
Amyloid na basi BRI proteinu (Abri). Jde geneticky podmíněnou poruchu zvanou Britská familiární demence, resp. dánská familiární demence, obě na společném podkladě amyloidosy vzniklé z transmembránového neuronálního proteinu (je přítomen za normálních okolností i mimo neurony, ale tam k prokazatelné deposici amyloidu nedochází. Na tkáňově úrovní je obraz deposice a distribuce analogický Alzheimerově nemoci. Amyloid je tvořen fragmentem abnormálního BRI proteinu zvaném ABri, kódovaném BRI2 genem. V případě dánské varianty se fragment, odlišné struktury, ale ze stejného výchozího proteinu BRI označuje Adan. V obou případech je deposice jak neuronální tak cévní.
Amyloid na basi prionů (APrP) . Priony (název je odvozen od jejich infekční a proteinové povahy) jsou normální komponentou celé řady tkání, zejména tkáně nervové. Jde o proteiny skládající se primárně z 253 aminokyselin, kódované jediným genem (množné číslo znamená existenci isoforem, daných posttranslačními modifikacemi: různou proteolytickou modifikací a/nebo glykosylací). Jsou produkovány jako sekretorické proteiny, resp. sialoglykoproteiny a jsou cíleny vesikulárním transportem do buněčné membrány. V neuronech putují axonálním transportem do výběžků. Maximálně jsou koncentrovány v oblasti synapsí. V buněčné membráně jsou zakotveny svým C terminálním koncem pomocí GPI (glykosylfosfatidylinositolu). Zpětným membránovým tokem při endocytose se dostávají do lysosomů a předpokládá se jejich recyklace mezi těmito kompartmenty. Jejich funkce je neznámá.
V lymfocytech se údajně zúčastní na procesu aktivace mitogeny. U myší s vyřazeným prionovým genem nebyly v průběhu pokusu zjištěny žádné podstatné funkční nebo struktuální změny CNS, projevovaly se u nich jen nevýrazné poruchy denního rytmu, poruchy spánku a poruchy paměti.
Molekula prionového proteinu se může stát patogenní (a infekční) posttranslační změnou své konformace, aniž by se změnila její primární struktura. Fyziologické priony mají minimální zastoupení beta struktury (beta struktura 3%, alfa helix 42%), zatímco u "patogenního prionu" je beta struktura strukturou dominantní (43%, proti alfa helixu 30%, viz též obr. 6). Změna konformace vyžaduje značné množství energie a není vyloučeno, že může být katalysována dalším faktorem, například proteinem typu chaperonu.
Změna konformace je pokládána za hlavní patogenní moment ve skupině fatálních neurologických onemocnění souhrnně zvaných jako prionová onemocnění (prion diseases), mezi něž v lidské patologii patří Jakobova-Creutzfeldova nemoc (a její nedávno popsaná nová varianta v souvislosti s epidemii bovinní spongiformní encefalopatie), Gerstmannův-Strauslerův-Scheinkerův syndrom, familiární fatální insomnie a kuru.
Změna konformace prionu má za následek vznik takřka absolutní resistence k proteolyse a následně progresivní akumulaci konformačně abnormální varianty prionu. Předpokládá se, že hromadění je dáno trvalou konversí nově syntetisovaných prionů na formy resistentní. Mechanismus poškození tkáně patogenními priony není znám. Poškozena je pouze nervová tkáň, přesto, že PrPsc jsou vždy v hostitelském organismu generalizované.použito
Byla prokázána generalizace mozkovou tkání z jednoho fokusu inokulace. Kumulace prionů je pro neuron fatální. Variabilní část kumulovaných prionů se mění na amyloid.
Tento proces se by se dal přirovnat k působení krystalků kysličníku křemičitého, který je rovněž nezničitelný a trvale tkáň během svých intracelulárních pasáží destruuje.
Terminologie: PrP = prionový protein, PrPc, případně PrPsen = normální, celulární prionový protein, sensitivní na proteolysu, PrPres , případně PrPsc = obecný název pro patologický prion s beta konformací, resistentní na proteolysu, původně isolovaný ze scrapie (ovčí prionové encefalopatie), deltaPrP = mutované formy prionů, PrPCJD, PrPGSS, PrPFFI = alternativní název pro priony isolované z uvedených prionových nemocí, PrPd případně jako souhrnný název prionu způsobujícího onemocnění, PNRP gen prionu.
Mechanismy vedoucí ke konversi prionu normálního na prion infekční jsou schematicky znázorněny probrány v následujícím textu.
1. Spontánní sporadické formy. Jde pouze o Jakobovu-Creutzfeldovu nemoc. Ostatní prionová onemocnění mají dědičný základ. Mechanismus vzniku sporadických forem není uspokojivě vysvětlen, pokud lze vyloučit exogenní původ a mutace v PRNP genu. Přítomnost polymorfismů M129M a E229K značí pouze zvýšenou predisposici, nikoliv kausální vztah (jde o polymorfismy). Přítomnost homozygotní mutace M129M se hodnotí jako vysoká predisposice pro infekci PrPCJD.
U sporadických případů je nutno brát v úvahu možnost fokálního vzniku patogenního prionu v jediném neuronu (na př. somatickou mutací PRNP genu) s následným šířením konformační změny neuronální sítí, jak bylo prokázáno experimentální inokulací patogenního prionu do mozků experimentálních zvířat. Za zmínku stojí, že nadměrná produkce prionů, indukovaná experimentálně transfekcí divokého genu, vedla k příznakům prionové nemoci s konversí části prionů na beta patogenní formu. Je však otázka nakolik se nadměrná produkce prionů může u sporadických lidských nemocí uplatnit. V úvahu připadá i jejich snížená degradabilita.
2. Infekční přenos patogenního prionu (t.j. prionem s beta konformací) z člověka na člověka byl prokázán jednoznačně u kuru (na podkladě kanibalismu), a v některých případech lidské Jakobovy-Creuzfeldovy nemoci (iatrogenní přenos). Existuje důvodné podezření, že některé ze sporadických atypických případů této nemoci, lišících se neuropatologicky od forem klasických (více amyloidních plak podobných kuru) jsou ve vztahu k bovinní spongiformní encefalopatii (u všech postižených byl přítomen predisponující polymorfismus M129V v homozygotní formě). Ve zvířecích chovech (hovězí dobytek, ovce) je za hlavní zdroj nákazy považováno infikované krmivo (např. masokostní moučka připravená z infikovaných zvířat), ale lze předpokládat i transplacentární přenos.
Infekčnost prionů je podle všech dosavadních experimentů vázána na přítomnost normálních prionů u hostitele. Podstatou infekčnosti a toxického působení patogenních prionů obecně je v dnešním pohledu interakce patogenního prionu s prionem normáním (tvorba dimerů) vedoucí ke změně konformace hostitelského prionu. Předpokládá se nezbytný stupeň homologie mezi oběma priony, zejména při mezidruhovém přenosu, který je jinak obtížný nebo nemožný.
Myši s vyřazeným PrP genem jsou resistentní k experimentální infekci priony. Samotná absence prionů však u nich nevede k prokazatené neurologické a neuropatologické poruše. Ukazuje se však, že případnou mezidruhovou barieru lze překonat způsobem inokulace, nejlépe přímou aplikací testované tkáně do mozku, zvýšením dávky či pasáží původně "cizího" prionu.
3. Vertikální, geneticky podmíněný, přenos mutace strukturálního PRNP genu je příčinou GSS, FFI a asi 10% případů JCN. Přenos je autosomálně dominantní s různou penetrancí. Manifestní nemoc vznikne i u nosiče jedné mutované alely prionového genu. Do dnešní doby bylo popsáno přes dvacet patogenních mutací a polymorfismů, které mají významný modifikující vliv. Není známo nic o možných homozygotních formách.
Předpokládá se, že mutantní priony jsou náchylné k spontánní patogenní beta konformaci. Pokud byl studován normální protein, produkovaný u nosičů druhou, nepostiženou (divokou) alelou, byla prokázána jeho nerozpustnost, ale sensitivita na proteolysu, což lze pokládat za potencionálně přechodný stav k patogenní beta konformaci. Tyto nálezy by mohly vysvětlit úspěšnost horizontálního (infekčního) přenosu inokulací extraktu tkáně mozku u většiny dědičně podmíněných případů humánních prionových onemocnění (JC, GSS, FFI) na myši a některé primáty. Takováto současná možnost vertikálního (genetického) a horizontálního (infenkčního) přenosu nemá v molekulární patologií geneticky podmíněných nemocí obdoby.
Předpokládá se rovněž, že mutace v prionovém genu by mohly predisponovat k infekci exogenním patogenním prionem. Jedním z důvodů pro tento předpoklad je existence jinak prokazatelně patogenních mutací u klinicky asymptomatických případů (nekompletní penetrance). To vše je důvod k úvahám, zda jsou priony výhradním patogenetickým agens. Uvažuje se o viru nebo o defektním proteinu chaperonového typu.
Patogenesa tkáňových změn u prionových nemocí. V presymptomatickém stadiu dochází ke kumulaci PRNPres v nervových výběžcích a v synapsích. Od určitého stupně kumulace nastane regrese neuronů s jejich zánikem nebo s různými histologicky patrnými degenerativními změnami (viz níže). Proces je lokalisován primárně v šedé hmotě, bílá hmota je postižena sekundárně jako následek neuronální degenerace. V dalším rozvoji změn dochází k různě vyjádřené precipitaci konformačně modulovaného prionu do deposit. Část prionů v placích může být přítomna ve formě amorfní, detegovatelné pouze imunohistologicky (primitivní, difusní plaky). Část plaků má priony uspořádané do amyloidních fibril s typickou tinkcí a ultrastrukturálním vzhledem. Z dosavadních pozorování se nedá jednoznačně uzavřít, zda se u geneticky podmíněných stavů podílejí na tvorbě amyloidních fibril výhradně mutantní priony, i když pro to některé nálezy svědčí. Množství amyloidu u základních variant prionových onemocnění silně kolísá. Tyto nálezy svědčí pro to, že fibrilární transformace nemusí být bezprostředním následkem konformačně změněných molekul prionů.
Patologický genotyp prionových nemocí se manifesuje třemi základními klinicko-patologickými jednotkami: Jakobovou-Creutzfeldovou nemocí, Gerstmannovou-Strausslerovou-Scheinkerovou nemocí a familiární fatální insomnií. Jakobova-Creutzfeldova nemoc, jako jediná z těchto tří má prokazatelně jak získaný (infekční), tak geneticky podmíněný vznik. Zbylé dvě byly popsány pouze jako geneticky podmíněné.
Jakobova-Creutzfelova nemoc. U dědičně podmíněné varianty byly doposud popsány mutace v kodonech 178 (tato v kombinaci s valinem v kodonu 129 na mutantní alele) 180, 200, 210, 232 prionového genu, a jedna inserce oktapeptidu na N-terminálním konci prionového proteinu. Neuropatologie je charakterisovaná masivní vakuolisací neuronálních výběžků v neuropilu (obraz t.zv. subakutní spongiformní encefalopatie) a zánikem neuronů a reaktivní astrogliózou (další detaily jsou v neuropatologických monografiích). Na pozadí masivně difusně zmnožených prionů (PrPres) se vyskytují nečetné prionové plaky jak primitivní (neamyloidní), tak amyloidní jen asi u 5-10% případů.
Gerstmannova-Strausslerova-Scheinkerova nemoc (doposud byly popsány mutace v kodonech 102, 105, 117, 145, 198, 217 prionového genu). Neuropatologií dominují četné amyloidní plaky, které jsou pro tuto variantu typické.
Fatální familiární insomnie (identická mutace v kodonu 178 jako u Jakobovy-Creutzfeldovy nemoci, ale v kombinaci s methioninem v kodonu 129 mutantní alely). Maximum neuropatologických změn je v některých jádrech thalamu. Prokazatelné jsou pouze silně zmnožené PrPres. Prionové plaky údajně chybí.
Pokud je mutace (považovaná nikoliv za kausální, ale pouze modulující, t.j. pouze v kombinaci s mutací patogenní) na 129. kodonu v homozygotním stavu (valin za methionin), je klinický průběh rychlejší.
Změny u onemocnění podmíněných priony nemusí být omezeny na centrální nervový systém. U ovčí prionové nemoci (scrapie), podobně jako u experimentálně (transfekcí) indukované zvýšené exprese prionů u myší je běžná těžká nekrotisující myositis bez deposice amyloidu. Zmnožené jsou však pouze PrPsen .
U lidské myositidy s inklusními tělísky, velmi časté ve starším věku (inclusion body myositis) jsou priony (zčásti PrPres ) spolu s AbetaP prokazatelné v příznačných kongofilních amyloidních inklusích. Může být samostatným projevem, ale byla popsána i v souvislosti s CJD.
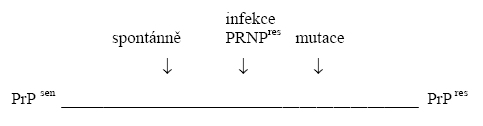
Amyloid na basi kalcitoninu (ACal). Kalcitonin je produktem C buněk thyreoidey. Je syntetisován jako preprohormon o 141 aminokyselinách. Dalšími úpravami - parciální proteolysou je odštěpena větší N-terminální a menší C-terminální část. Výsledný hormon má 32 aminokyselin. V amyloidu byla prokázána jak kompletní forma kalcitoninu, tak některé delší nekompletně proteolyticky upravené varianty. Amyloid na basi kalcitoninu se vytváří pouze v nádorově transfornované tkáni (t. zv. medulární karcinom thyreoidey). V nenádorové štítné žláze nebyl doposud prokázán. Tím se liší od ostatních popsaných endokrinních amyloidů. Jako všechny endokrinní amyloidosy je však omezen na okolí nádorově transformované endokrinní buňky.
Ukazuje se, že C buňky jsou sice hlavním, ne však jediným buněčným typem produkujícím kalcitonin. Jeho produkce byla prokázána i v hepatocytech.
Kalcitonin je kodován genem patřícím do skupiny homologních genů, do které patří též gen betaCGRP (calcitonin gen related peptide) a gen amylinu. Gen kalcitoninu produkuje dvě sestřihové varianty: kalcitonin a t.zv. alfaCGRP s odlišným biologickým účinkem (jde o neurotransmiter, blíže viz doporučená literatura). Jak kalcitonin tak jeho sestřihová varianta (alfaCGRP) jsou produkovány buňkami medulárního karcinomu. Gen betaCGRP je vysoce homologní s předchozím genem. Gen amylinu koduje peptid amylin (viz níže). Oba CGRP mají charakter neuropeptidů a mají vasodilatační účinek. Spolu s amylinem se účastní regulace metabolismu glukosy s účinkem protichůdným insulinu. V C buňkách thyreoidey a v nádoru z C buněk je nejvíce exprimovaná produkce kalcitoninu a zčásti jeho isoformy alfaCGRP. Exprese zbylých dvou genů je minimální nebo je utlumena zcela.
Amyloidosa na basi medinu (AMed). Jde o získanou amyloidosu topicky omezenou, podle současných poznatků, na svalovou vrstvu artérii. Medin (název podle výskytu v medii) je částí molekuly laktadherinu, který je produkován hladkými svaly stěny cévní, ale i dalšími buňkami mimo cévní stěnu, na př. epitelem mléčné žlázy. Amyloidní fibrily složené z tohoto fragmentu se vyskytují takřka ve 100% v medii aorty starých lidí. Zvýšené množství bylo prokázané ve stěně cévní u temporální arteriitidy. Jeho význam v patologii arteriální stěny je pravděpodobně velmi malý. Důsledky jeho deposice však nebyly doposud studovány.
Amyloid na basi laktoferinu (ALac). Laktoferin je produkován slznou žlázou a je kvantitativně výrazně zastoupeným proteinem slz. ALac byl prokázán v rohovce jako průvodní znak oční trichiasy, a některých dalších očních onemocněních, tedy jako sekundární fenomén. Predisposici k vzniku amyloidu je mutace v příslušném genu.
Amyloid na basi semenogelinu I (ASgI) je jedním z amyloidů provázející proces stárnutí. Jde o amyloidosu vesiculae seminales (semenných váčků). Zmíněný protein je hlavní komponentou sekretu žlázky. Může být provázen laktoferinem. Afekce je zcela lokální a je zcela nezávažná.
Amyloid na basi keratoepithelinu (AKer). Je podstatou jedné z forem geneticky podmíněných dystrofii rohovky. Deposice amyloidu není vázána na zánětlivé procesy oka. Je podmíněna mutací v genu TGFbetaI (indukovaném TGFbeta), který kóduje protein keratoepithelin. Přenos je autosomálně dominantní. Keratoepithelin byl prokázán jako průvodní komponenta jiných amyloidos oka.
Amyloid na basi cytokeratinu (AKer) byl prokázán imunohistochemicky v kožních lesích typu lichen amyloidosus a v t.zv. makulární amyloidose, což jsou autochtonní kožní onemocnění (ne tedy příznaky generalizovaných amyloidos !). Jde o tzv. primární kožní amyloidosu. Dále je amyloid na basi cytokeratinu prokazatelný jako lokální sekundární proces při některých kožních nádorech (trichoepiteliom, basaliom, Bowenova dermatosa a některé další) - sekundární kožní amyloidosa. Dostupné výsledky jsou doposud pouze z imunohistochemických studii. Jejich společným jmenovatelem je, že jde o typy cytokeratinu (nejčastěji CK 5) patřící do skupiny keratinů basických (skupina II).
Poznámka. Buněčná patologie této amyloidosy se liší od všech předchozích tím, že jde o extracelulární deposita amyloidogenního proteinu na basi cytoskletálního proteinu, který není normálně secernován. Jediné srovnání je možné s amyloidem na basi gelsolinu (viz shora), což je protein působící v cytosolu, účastnící se na regulaci aktinu, u kterého však byly prokázány sestřihové sekretorické varianty. V případě sekundární kožní amyloidosy je diskutován vznik ze zanikajících nádorových buněk.
Amyloid na basi amylinu (AIAPP). Amylin (islet amyloid polypeptid) je peptidový hormon produkovaný a secernovaný spolu s insulinem beta buňkami pankreatických ostrůvků. Vyskytuje se i v některých dalších úsecích GIT a v buňkách periferního endokrinního systému. Je tvořen peptidem o 37 aminokyselinách, uvolněným z prekursoru o 89 aminokyselinách. Peptid je kosegregován v sekrečních granulích s insulinem a společně s ním přechází do krve. Za normálních okolností je dále proteolyticky zpracován na menší peptidy. Jde o produkt jednoho ze skupiny t. zv. kalcitoninových genů, spolu s kalcitoninem (a jeho sestřihovou variantou alfaCGRP) a s t.zv. beta CGRP (viz shora). Svým účinkem je protichůdný insulinu (tlumí syntesu glykogenu, vstup glukosy do buněk, stimuluje degradaci glykogenu). Fysiologicky se vyskytuje v lysosomech B buněk ostrůvků spolu s insulinem. Jde pravděpodobně o výraz regulace sekrece lysosomálním systémem (krinofagie, viz prvý díl skript).
Deposice amylinového amyloidu je pouze lokální. Probíhá velmi často v závislosti na věku a subklinicky. Kritickým amyloidogenním faktorem se zdá být nadměrná stimulace sekrece beta buněk. Snad proto je deposice amyloidu vystupňovaná nejvíce u diabetu typu II a je pravděpodobné, že k zániku beta buněk za tohoto stavu přispívá (viz níže). Podle doposud ojedinělých studií začíná deposice amyloidu intracelulárně jednak volně v cytosolu, jednak v depositech ohraničených membránou. Byly popsány komunikace i.c. deposit s extracelulárním prostorem. Naprostá většina amyloidu je deponována extracelulárně. Velmi často je amyloidogenese vystupňována u insulinomů z beta buněk. Amyloid se skládá z celého peptidu i jeho fragmentů. Kritická amyloidogenní (beta) struktura je mezi 2O. a 30. aminokyselinou.
Toxicita amylinu byla prokázána v tkáňových kulturách lidských a krysích Langerhansových ostrůvků, obohacených exogenně přidaným amylinem. Toxicita byla vázána na fibrilární agregaci amylinu do amyloidních fibril v mediu. Podobně toxicita testovaných amylinů různých živočišných druhů byla vázána na schopnost jejich fibrilární agregace.
Amyloid na basi insulinu (AIns) spontánně vznikající nebyl doposud u člověka popsán. Lokální amyloid z insulinu však byl prokázán v místech aplikací tohoto hormonu u diabetiků typu I (iatrogenní amyloidosa). Spontánní výskyt amyloidu na basi insulinu byl popsán u některých hlodavců (Octodon degus).
Atriální srdeční amyloidosa na basi atriálního natriurického peptidu (ANP). ANP amyloidosa, zvaná též ANF (dle atrium natriuretic factor) je velmi častým nálezem v srdečních síních (myokard komor není postižen) u starších lidí. Podle některých sestav se vyskytuje u lidí v šestém deceniu v 67%. Jde o fibrilární agregaci síňového natriurického hormonu (atrial natriuretic peptide - ANP) a to jak prohormonu, tak i jeho fragmentů, které z něho normálně vznikají a které představují aktivní formu hormonu.
Je syntetizován jako prohormon (proANP1-126) a je proteolyticky upraven na C-terminální peptid (ANP99-126, zvaný kardiodilantin) a N-terminální peptid (ANP1-98). Oba tyto peptidy byly v atriální amyloidose prokázány. Spolu s atriálním natriurickým hormonem je v malém množství spoluprecipitován do amyloidu i t.zv. mozkový natriurický peptid (BNP-brain natriuretic peptide), který je za normálních okolností syntetizovaná v malém množství i v síních srdce. Drobná deposita amyloidu byla prokázána intracelulárně v kardiocytech síní, většina deposice je však extracelulárně v intersticiu, zčásti subendokardiálně a v cévách síní. Jde o lokalizovaný amyloid, deponovaný v nejbližším okolí produkující buňky. Síňová amyloidosa nemá definovatelný klinický korelát. Predispozicí a akcelerujícím faktorem se může být hypersekrece hormonu při srdečním selhávání.
Amyloid v adenohypofyse. Deposice amyloidu v prostoru adenohypofysy je velmi častý nález, úměrný věku. Rovněž často je amyloid deponován v hypofysárních adenomech o různých sekrečních profilech. Proces deposice amyloidu probíhá subklinicky. Charakteristika amyloidního proteinu je technicky obtížná pro malý rozsah změn. Nedávno byl identifikován jako prvý amyloidogenní hormon prolaktin (resp. jeho fragmenty) v senilním amyloidu hypofýzy . Jde tedy o amyloid na basi prolaktinu (APro)
V řadě případů existuje pouze empiricky prokázaná amyloidosa na podkladě fysikálního průkazu.
Z toho důvodu je existuje stále čistě deskriptivní termín senilní orgánová amyloidosa, zahrnující deposici amyloidu v závislosti na věku v adenohypofyse (výjimka viz výše), v kůře nadledviny (v obou lokalitách je popisována diskrétní jak intra- tak extracelulární deposice. V žilách portální oblasti byl u starých lidí nalezen v 37% amyloid zčásti imunohistochemicky neurčený, zčásti na basi TTR.
Výrazná deposice amyloidu v závislosti na věku probíhá v oblasti epitelu plexus chorioideus. Amyloidogenní protein však není znám. Proces doprovází intracelulární deposice argyrofilních, kongofilních a dichroických vláknitých struktur, blízkých párovým helikálním filamentům v neuronech (viz NTF u Alzheimerovy nemoci), zvaných v této lokalizaci Biondiniho tělíska. Tato tělíska byla nalezena i v řadě endokrinních buněk.
Deposice amyloidu u všech těchto stavů probíhá subklinicky.
Část amyloidogenních proteinů ze skupiny senilní amyloidosy byla již určena, jako aortální intimální senilní amyloidosa na basi Apo A1 (viz výše), nebo senilní kardiální amyloidosa na basi TRR (viz výše). Nutno připomenout poměrně významnou, byť subklinicky probíhající cerebrální amyloidosu na podkladě AbetaP (viz výše).
Ve studovaných sériích šlo o incidenci řádově desítek procent naznačující, že jde o projev velmi častý, jehož intensita však může kolísat. Tento základní proces může být akcelerován dalšími faktory. Patogeneticky lze tuto formu amyloidosy těžko hodnotit. Amyloidogenní proteiny mohou být a jsou v různých orgánech různé, takže lze na tento stav pohlížet jako na spontánně probíhající biochemicky odlišné orgánově specifické amyloidosy, tedy jako na senilní "mnohočetnou amyloidosu " (každá z amyloidos na basi jednoho typu proteinu), mnohočetný proces, nejspíše způsobený procesem stárnutí (AApoA1 v aortě, AbetaP v mozku, amyloid na basi prolaktinu v adenohypofyse, na basi TTR v myokardu, semenogelinový amyloid v semenných váčcích, neznámý amyloid v plexus chorioideus). Patří sem i doposud ne zcela jednoznačně definovaný amyloid v chrupavce, v synovii kloubní a v meziobratlových ploténkách prokazatelný ve vysokém procentu ve stáří, zejména v degenerativních procesech typu arthrosy (studie z poslední doby prokazují vznik z Apoproteinu AI, který je produkován chondrocyty). Nutno připomenou i kombinovanou ATTR a AApoAIV amyloidosu - viz ApoAIV amyloid). S touto senilní "polyamyloidosou" samozřejmě kontrastují amyloidosy způsobené isolovanou poruchou obratu jednoho určitého amyloidogenního proteinu.
Poněkud stranou stojí senilní "monoproteinová" generalizovaná TTR amyloidosa, odlišující se od isolované senilní TRR amyloidose myokardu a od dědičně podmíněné TRR amyloidosy. V každém takovém případě je však nutno na dědičný původ pomýšlet.
Část neurčených amyloidos však representuje klinicky manifestní poruchy. Jde o amyloid v osteoarthrotických lesích (i když zde jde spíše o sekundární, průvodní jev).
Povšechné amyloidosy získané. Amyloidogenním proteinem je protein normální. V některých případech však může hrát roli polymorfismus.
Povšechné amyloidosy podmíněné geneticky. Amyloidogenní jsou mutantní proteiny, nebo jejich fragmenty.
Zde jsou zmíněny pouze stavy s výrazným postižením určitého orgánu, tedy na pozadí generalizace procesu amyloidosy
renální postižení : je běžné u získaných generalizovaných amyloidos.
Familiární postižení je typické pro amyloidosu na basi:
hepatání postižení: je poměrně výjimečné u získaných forem (AL, ATTR).
Familiární těžké postižení se ukazuje být příznačné pro amyloidosu na basi:
neuropatické forma (periferní neuropatie): přichází v úvahu u získaných forem.
Z dědičných je to především amyloidosa na basi:
amyloidosa myokardu: je častá u získaných forem na basi
amyloidové opacity ve sklivci : deposice amyloidu z transthyretinu
amyloid v rohovce:
kožní amyloidosa : Amyloidosy omezené na kůži jsou poměrně časté. Dělí se na primární (bez vyvolávající lése a sekundární (při primární kožní lési) . Bohužel, mnohdy byl amyloid prokázán pouze základními technikami, aniž by byl dále studován immunohistochemicky nebo biochemicky. Pokud byl ložiskový primární amyloid studován, šlo o imunoamyloid při infiltraci ložiska plasmocyty. U ložiskových amyloidos sekundárních to byl velmi často cytokeratiin, ale řada studii poukazuje na apoprotein E, u kterého je nutno brát v úvahu, že je syntetisován keratinocyty). Jako součást generalizovaných amyloidos (získaných, geneticky podmíněných) je kůže postižená v řadě případů - u immunoamyloidos, u SAA amylodiosy, u amyloidos na basi mutantního transthyretinu.
amyloidosa s postižením mozkových cév (dříve tzv. kongofilní angiopatie): (včetně neurologické symptomatologie) - neurologicky závažné amyloidosy (mimo prionosy, které jsou svým základním projevem odlišné).
Dvě z těchto formy nemají viscerální postižení (klasická biopsie není tedy přínosná):
amyloid aortální (ve stáří)
Tato schémata vycházejí z literárního přehledu. Vzhledem k variabilitě tkáňových změn u amyloidos nemusí pokrývat vyčerpávajícím způsobem celou problematiku diferenciální diagnosy
závěr - identifikace amyloidu versus diagnosa amyloidosy
Identifikace amyloidu jakožto betafibrily není v zásadě žádný zásadní problém (vazba Kongo červeně s indukcí dvojlomu a dichroismu, ultrastrukturální průkaz fibril). Velmi obtížná je identifikace typu amyloidosy, která naráží v praxi na obtížnosti immunohistochemické detekce stavebního kamenu amyloidu in situ v rutinních parafinových řezech. Optimálně byl měl být každý amyloid vyšetřen biochemicky po extrakci nefixovaného vzorku, např. kryostatového řezu (ideálně po selekci od ostatních komponent mikrodisekcí) a podroben sekvenační nebo hmotnostní analýze. Do jisté míry je toto možné i v případě standardního parafinového vzorku tkáně. Tím by byl ideálně stanoven molekulární typ. Po té by mělo následovat hodnocení procesu, který k amyloidose vedl, včetně toho, zda jde o získaný, či geneticky podmíněný proces, případně charakter fragmentů amyloidogenního proteinu (stupeň proteolytické degradace u získaných forem, účast mutantního proteinu ve srovnání s divokým proteinem u geneticky podmíněných amyloidos, které mají dominantní přenos)
V hodnocení amyloidos nutno brát v úvahu :
U klasické sekundární AA amyloidosy přichází ve výjimečných případech v úvahu objasnění genetického přenosu v rodinách. Primárně geneticky podmíněná AA amyloidosa nebyla doposud jednoznačně prokázána. Rozhodující se stále ukazuje být genetický přenos její primární příčiny, stručně zmíněné níže.
Okrajovou záležitostí (i když biologicky velmi zajímavou a v budoucnosti velmi pravděpodobně velmi významnou) může být nejen stanovení příměsi nefibrilárních (nekongofilních) komponent kritického amyloidogenního proteinu (zejména v případě AL amyloidosy, viz výše), ale i stanovení molekulárních variant deponovaného proteinu a tím se přiblížit mechanismu prefibrilární a terminální fibrilární agregace a role buňky samotné v mechanismu amyloidosy. V souhrnu jde o sekvenci: amyloid-specifikace stavební molekuly, popřípadě specifikace mechanismu její agregace
Pouhé konstatování, že v bioptickém (nebo pitevním) vzorku je prokazatelný amyloid není dostatečné. Jde o diagnosu na úrovni biofysikální, nikoliv na úrovni biologické. Pouze diagnosa na úrovni biologické má zásadní výpovědní hodnotu, neboť vypovídá nejvíce o specifické patogenese stavu. V tomto smyslu jsou klinici zcela závislí na expertise patologa, resp. molekulárního patologa nebo biochemika.
identifikace neznámých amyloidů
Nové typy amyloidu, s doposud neznámým výchozím amyloidogenním proteinem se identifikují biochemickou cestou, která zahrnuje solubilisaci proteinů ve studovaném vzorku, jejich elektroforesu, isolaci kritického proužku a sekvenování nebo hmotnostní analýza. Optimální je isolace amyloidového deposita mikrodisekční technikou (odstranění nespecifických proteinů). Po stanovení jeho primární struktury pak může být protein určen srovnáním s doposud známými proteiny podle příslušných sekvenčních databází, zjištěn jeho gen a podroben sekvenční analýze.