This text represents commentary notes to lectures and seminars on cytoplasmic topics of the Introductory Course of Cell Biology for the 1st year M.D. program students of the Charles University in Prague - First Faculty of Medicine. The informational content of this text, lectures and seminars represent minimal knowledge requirements in these topics.
The underlying and integrating idea embedded into this text is the one stating: "Cellular proteins must be properly synthesized and folded but also properly and correctly targeted into cellular compartments, in order to function optimally." It was our intention to demonstrate the complexity of cytoplasmic cellular organelles both through their structural organization as well as biochemical principles of their biogenesis. We consider this integrative approach as an essential prerequisite for understanding of complex cellular/biologicalphenomena.
It is our firm belief that the fundamental principles of human pathology as well as efficient causal biological therapy can be impartibly understood only through comprehensive lecturing of basic principles of cell biology. Therefore, we included brief introductory notes to emphasize the importance of particular biological topics for the students´ future studies at the Faculty. We strongly encourage the students to actively search for additional information during the forthcoming courses (biochemistry, microbiology, immunology, pathology as well clinical medicine).
The text includes active links to Figures and Tables in the available on-line version of the Molecular Biology of the Cell textbook and other lecturing texts prepared for this course; additional Figures are appended at the end of this document and are linked from the text. For those interested, we included a list of elective literature represented by recent review articles in peer-reviewed scientific journals. Students are kindly asked not be afraid of the complexity of these texts and, if not now, return to these texts or their more recent versions during later years of their studies. Full text versions of these elective reviews should be accessible from the computers connected to the Faculty's computer network.
We are grateful for any inspiring and critical, negative included, comments related to this text and hope that it will serve the above-mentioned concept.
This document must not be used for other than academic purposes.
Autumn 2008 - Milan Elleder a Jakub Sikora
3D - three dimensional; Aa - amino acid; ATP - adenosine triphosphate; ATPase - ATP synthase; Cdk - cyclin dependant kinase; CMA - chaperone mediated autophagy; COP - coatomer protein; DNA - deoxyribonucleic acid; EDEM - endoplasmic reticulum degradation-enhancing α-mannosidase like; ELS - endosomal lysosomal system; ER - endoplasmic reticulum; ERAD - endoplasmic reticulum associated degradation; ERAF - endoplasmic reticulum associated folding; ESP - early secretory pathway; gDNA - genomic DNA; GPI - glycosylphosphatidylinositol; GTP - guanosine triphosphate; GTPase - guanosine triphosphatase; LAMP - lysosomal associated membrane protein; LIMP - lysosomal integral membrane protein; LMP - lysosomal membrane protein; LRO - lysosomal related organelles; LSD - lysosomal storage disorders; M-6-P - mannose-6-phosphate; M-6-PR - mannose-6-phosphate receptor; MALS - macroautophagy lysosomal system; mRNA - messenger RNA; mtDNA - mitochondrial DNA; OXA - mitochondrial transmembrane translocator OXPHOS - oxidative phosphorylation; PTS - peroxisomal targeting sequence; Rab - GTPase (rabbit-like); RER - rough endoplasmic reticulum; RNA - ribonucleic acid; rRNA - ribosomal RNA; SER - smooth endoplasmic reticulum; SNARE - soluble n-ethyl maleimide sensitive attachment proteins receptor; SRP - signal recognition particle; TIM - translocase of the inner mitochondrial membrane; TOM - translocase of the outer mitochondrial membrane; tRNA - transfer RNA; UDP - uridine diphosphate; UPR - unfolded protein response; UPS - ubiquitin proteasome system
Biogenesis refers to a principle when living organisms, individual cells included, develop from living matter, i.e.. cells originate from pre-existing cells. Cytoplasm (all extra nuclear cellular components) comprises all membranous organelles and cytosol (aqueous fluidic environment of the cell). Cytosol harbors a number of complex and ordered non-membranous (proteinaceous) systems such as cytoskeletal scaffold, protein synthesis machinery or components of various intracellular metabolic or signaling pathways (Alb. 12-1 (on-line)). It is worth stressing that the overall size, surface and nuclear/cytoplasmic volume ratio are not constant throughout the cell cycle and may vary with differentiation stage or functional status of the cell.
Dynamic and continuous remodeling and compartmentalization of the eukaryotic cellular interior is essential and indispensable for normal cellular functionality and viability. The primary function of biological membranes is to maintain the integrity and to delimit divergent individual organellar compartments. Additionally, biological membranes (hydrophobic phases) (Alb 10-1 (on-line), 10-12 (on-line))either through direct contacts or specialized membrane proteins/protein complexes, mediate communication among cellular compartments. Therefore, it is not surprising that sustenance of such a continuously changing system with various, but inherent disequilibria and gradients, requires energy input.
As introduced above, the dynamic biogenesis of membranous organelles occurs on the basis of pre-existing structural substrate, which ontogenetically originates from the oocyte's cytoplasm, i.e. is of maternal descent, non-nuclear DNA (mitochondrial DNA - mtDNA, see below) included. The ongoing renewal of the membranous compartments in every eukaryotic cell requires precise targeting of their primary constituents (proteins, lipids or carbohydrate derivates) either from sites of synthesis or via regulated re-cycling.
Individual proteins are coded by genes in the nuclear/genomic DNA (gDNA). This information is further transcribed into the corresponding mRNA molecules, which consequently serve as templates for protein synthesis (translation into the sequence of amino acids) on the ribosomes. These newly emerging protein molecules become folded (see below and also Gene Expression)) and may get extensively posttranslationally modified (e.g. glycosylated, proteolytically cleaved etc.). Moreover, proteins must be coordinately transported (targeted) to specific cellular compartments or sub-compartments (membranous or aqueous in principle). Proteins with extracellular function are exocytosed from the cell (see below).
The cellular processing and exact protein targeting is regulated by the presence of either signal sequences in the primary structure of newly synthesized proteins or by 3D signal patches in the tertiary structure (Alb 12-8 (on-line))of the folded proteins. The difference between the signal sequences and signal patches is fundamental.
Signal sequences are preferentially N-terminally located stretches of 20-25 amino acids with predominating hydrophobic properties (Alb Table 12-3 (on-line)). In case of signal sequences, it is the primary amino acid sequence, which specifically determines the targeting of newly synthesized protein molecule to the proper cellular compartment (endoplasmic reticulum, nucleus, mitochondria or elsewhere) or sub-compartment. On the contrary, 3D signal patches emerge (become recognizable) after proper protein folding (see below).
The proteosynthesis of all eukaryotic proteins starts on free cytosolic ribosomes or polyribosomes (Alb 12-6 (on-line), 12-37 (on-line), see also Gene Expression). As mentioned above, the presence or absence of the specific N-terminal endoplasmic reticulum import signal sequence determines whether the protein will be co-translationally translocated into the cisternae of rough endoplasmic reticulum, posttranslationally processed in Golgi apparatus and further directed into the endosomal/lysosomal system or exocytosed (see below).
Newly synthesized proteins lacking N-terminal endoplasmic reticulum import signal sequence remain in the cytosol or are targeted to nucleus, mitochondria or peroxisomes depending on the presence of different types of signal sequences or signal patches (Alb Table 12-3 (on-line)) (for details see below and also Gene Expression)
Proteostasis can be defined as a complex system of pathways involved in cellular and biochemical homeostasis of proteins. These pathways include complex mechanisms of protein synthesis, folding, modification, targeting, as well as degradation in the cell.
Theoretically, protein translation results in a linear polypeptide chain, which is for obvious reasons energetically unstable. Newly synthesized proteins adopt energetically favorable conformation (native structure), meaning that the hydrophobic residues become buried in the insides of proteins and hydrophilic residues are, on the contrary, exposed on the solvent-accessible surfaces. The formation of the native structure further includes stabilization through hydrogen bonds, van der Waals' and electrostatic interactions and sometimes also by covalent bonding. It can be assumed (based both on experimental as well theoretical data) that the native state of a protein represents the most stable conformation under physiological conditions.
The term protein folding describes a formation of the energetically most favorable (lowest free energy) secondary and tertiary structure of the molecule (Alb 6-82 (on-line)) . On the contrary, the acquisition of the quarternary protein structure is defined as oligomerization or protein complex formation, although the physical nature of these processes is but an extension of folding (Alb 6-79 (on-line)). The exact difference between oligomerization and protein complex formation is sometimes nebulous. Subunits of oligomers are permanently associated and are handled and degraded by the cell in a unitary fashion; protein complexes are more dynamic with considerably higher freedom of processing.
The prerequisites necessary for acquisition of native protein structure seem to be an inherent property of the amino acid sequence itself. Nevertheless, it has been postulated that it is impossible for a protein to randomly sample all potential conformations (conformer states) in order to reach the most stable one. Currently accepted model of protein folding suggests that proteins follow a declining free energy landscape to gain the necessary energetic minimum of the native state (for additional details lectures and seminars). It seems plausible to assume that larger protein molecules fold in modular way. Folding thus occurs in different, largely independent regions or domains of the protein, which subsequently form the correct overall structure.
Folding of a newly synthesized protein can start once the N-terminus emerges from the ribosomal channel, be it either free cytosolic ribosomes/polyribosomes or ER associated ribosomes (Alb 6-81 (on-line)). Even though nascent protein (Alb 6-80 (on-line)) molecule can acquire the native conformation on its own (self-catalytically), this mechanism is most unlikely in the intracellular, aggregation prone environment that is packed with other proteins. It is the primary function of complex networks of folding enzymes and chaperones to catalyze transient folding steps of newly synthesized proteins, prevent unwanted protein/protein interactions or appearance of misfolded (potentially aggregative) protein variants (Alb 6-83 (on-line)). Chaperone can be defined as a protein aiding folding and assembly of other proteins (see above for the impartible association of folding and oligomerization). Chaperones are not integrated into final functional protein assemblies.
As can be derived from the above stated, proteins, including those destined for membranous compartments such as mitochondria, are synthesized on free cytosolic ribosomes/polyribosomes, and are handled by cytosolic chaperone protein machinery. Nevertheless, final folding of mitochondrial proteins occurs in mitochondria (see below). Proteins synthesized into ER cisternae or ER membranes are aided by the ER associated folding (ERAF) network (see below). Interestingly, as has been recently demonstrated, some peroxisomal proteins are transported through peroxisomal membrane in a folded state from cytosol.
For nuclear proteins refer to Gene Expression section.| Chaperone assisted protein folding, oligomerization, protein complex formation and proteostatic regulation are indispensible for normal cellular function. The processive sequence: proteosynthesis/compartment specific folding/protein assembly is employed by virtually every eukaryotic protein. It is advisable to keep this molecular sequence in mind while studying complex protein processing pathways. Additioanaly, it should not be surprising that such an amount of human diseases is associated with protein misfolding and aggregation (see below). |
In principle, there are several basic types of protein transport mechanisms - (i) transport through nuclear pores (see Gene Expression, Alb 12-10 (on-line)); ii) vesicular transport - transport in the lumina or membranes of transport vesicles; (iii) trans-membrane transport and membrane contacts; and (iv) non-vesicular transport through cytosol (transport of individual protein molecules or protein complexes by molecular transport systems directly associated with cytoskeleton or with non-membraneous cytosolic components).
This text primarily focuses on the vesicular transport and trans-membrane and membrane contact transport.
In the most general point of view, cellular proteins can be divided into two major groups based on their capacity to exist or effectively interact with hydrophobic phases (lipidic biological membranes) - water soluble and membrane proteins (Alb 10-17 (on-line))The capacity of a protein to either preferentially function in aqueous media (water soluble) or as an integral membrane (single or multiple trans-membrane) or membrane associated (via lipidic anchor such as glycosylphosphatidyl inositol - GPI (Alb 12-57 (on-line)), additional adaptor protein or bound through fatty acid) predetermines the mode of the protein's intracellular trafficking. Water soluble proteins can be transported as intraluminal vesicular cargoes; membrane proteins must be synthesized and remain in association with the hydrophobic environment of the biological membrane (for details and examples see below).
There are two types of transported cargo molecules (e.g. proteins) and two modes of their transport - (i) compounds destined for release into the "free" space (other intracellular compartments or extracellular space) - these are transported in the lumen of the transport vesicles (luminal transport) or bound to the luminal domain of their receptor which is embedded into the transport vesicle membrane; (ii) compounds destined for membrane insertion (e.g. proteins or lipid derivates). These are incorporated into the membranes of the transport vesicles.
Vesicular transport encompasses the following major intracellular cargo handling pathways - secretory pathway (exocytosis), endocytosis and transcytosis, processes which connect the intracellular compartments with cytoplasmic membrane and extracellular space (for details see below).
Vesicular transport is one of the hallmarks of living eukaryotic cells. The general principles of vesicular transport encompass the principle of vectoriality and specificity of shuttling between donor (departure) and target/acceptor (arrival) compartments through the following steps 12-7 (on-line), 13-2 (on-line)): budding (formation of transport vesicles), detachment from the donor compartment, targeted transport through cytosol along the cytoskeleton (mainly microtubules), specific docking/tethering/porus formation (association with the target compartment), and fusion with the acceptor compartment.The final step is the release of the luminal contents either into the target compartment or extracellular space and/or incorporation of the membrane proteins into the biological membrane of the acceptor compartment.
Budding (membrane bending) of biological membranes can be attained by extra-membranous protein scaffolds composed of multimerised protein subunits (Alb 13-7 (on-line))Proteins typically forming vesicular scaffolds are clathrin (Alb 13-41 (on-line)), coatomer proteins (COPs) or caveolin, which forms caveolae (coated pits) (Alb 13-42 (on-line)). These proteins can be differentially employed within various cellular compartments (Alb 13-5 (on-line)) or may constitute local membrane-associated accumulations (typically clathrin in coated pits functional in receptor mediated endocytosis Alb 13-46 (on-line) - see below). The interaction of these scaffold proteins with biological membranes is exerted through a number of various adaptor proteins or through cytoplasmic tails of transmembrane cargo binding receptors (Alb 13-8 (on-line)). Scaffold proteins can be recycled back to the donor compartment.
The second major mechanism used for membrane shape remodeling is the active redistribution of different types of lipid molecules in outer and inner sheets of biological membranes. This mechanism is used for formation of flattened endoplasmic reticulum and Golgi stacks with relatively sharp peripheral curvature or in dynamically remodeling tubulovesicular projections of late endosomes.
| For some selected topics of the structural clathrin assembly involved in membrane budding see review by Ungewickell and Hinrichsen, 2007. |
Detachment of the budding vesicles is primarily mediated by dynamin and related proteins, which function as molecular stranglers (Alb 13-8 (on-line), 13-9 (on-line)). Dynamin and dynamin-like proteins are also active in mitochondrial and peroxisomal network remodeling via exertion of fission events (see below).
Cytoskeletal associated vesicular trafficking - the movement of transport vesicles is an oriented (vectorial) and regulated process. Similar fundamental transport principles are utilized by all membranous cellular cargoes, including organelles such as mitochondria or peroxisomes. The interaction between the vesicular cargo and cytoskeleton (especially microtubules) is mediated by molecular motor proteins (dynein and kinesin) and their associated partner adaptor proteins (Alb 16-63 (on-line)). The overall vesicular transport within the cells is immense. It is worth noting that in polarized cells (e.g. neurons), the cytoskeleton mediated vesicular trafficking represents a prerequisite for homeostasis maintenance and cellular viability.
| Neuronal axons harbor a continuous bidirectional flow of membranous and non-membranous (large protein complexes) cargoes along their cytoskeletal scaffold, and are also involved in a wide variety of endocytic/exocytic, synaptic and autophagosomal cycling events associated both with anterograde (towards the axonal periphery) and retrograde (towards the neuronal soma) trafficking. Molecular pathology of axonal vesicular transport has been repeatedly demonstrated as major contributor to pathogenesis of number of neurodegenerative disorders including Alzheimer´s disease. For review on human axonal transport defects see Roy et al., 2005. |
Molecular coats of transport vesicles - transport vesicles carry a number of membrane proteins that function within the complex targeting network of specific intracellular vesicles. These proteins function as molecular tags of both donor and acceptor compartments as well as labels of luminal contents carried by the vesicle. The complex set of these proteins and glycoproteins establishes the so-called coat of the vesicle and represents its molecular identity. There are two major groups of membrane or membrane associated proteins which mediate this continuous process of vesicular shuttling and recognition - Rab and SNARE proteins (Alb 13-11 (on-line), 13-12 (on-line), 13-15 (on-line)). For review about the complexity of the vesicle recognition and shuttling network see Cai et al., 2007.
Specific docking and membrane fusions with acceptor compartment - Selective fusion of transport vesicles with acceptor (arrival) compartments is assured by specific interactions of complementary SNARE proteins (Alb 13-12 (on-line)). The membrane protein and successive membrane fusions are regulated by GTPase activity of Rab proteins (Alb 13-14 (on-line)). Regulated exocytosis (see below) of tethered (pre-docked) secretory vesicles is a transient and rapid process triggered by a specific signal.
Transport of structural proteins, enzymes (including enzyme subunits and assembling proteins), and membrane components into mitochondria and peroxisomes occurs by transport through cytosole and later through limiting membranes of these organelles. These processes are essential for biogenesis of these cellular compartments (see below). Mitochondrial proteins (coded by nuclear gDNA) and majority of peroxisomal proteins (see below) are devoid of the endoplasmic reticulum signal sequences and are therefore not translocated to the ER cisternae and are thus fully synthesized on free cytosolic ribosomes. Their proper cellular targeting is assured by specific organellar signal sequences (Alb 12-3 (on-line)). As briefly mentioned above, a fraction of proteins crosses the limiting peroxisomal membrane in a folded state. On the contrary, majority of mitochondrial proteins are transported by the mitochondrial transmembrane translocators (TOM, TIM and OXA) in an unfolded state (see below).
Early secretory pathway (ESP) represents a functionally compound compartment comprising endoplasmic reticulum (ER), pre-Golgi intermediates, also referred to as vesicular tubular clusters or ER/Golgi intermediates, which shuttle cargo between ER and Golgi apparatus. The transfer of cargo between ER and Golgi is oriented both from ER to Golgi as well as retrogradely from Golgi to ER, assuring recycling and retention of ER resident proteins within ER. These two transport directionalities are associated with different coatomer protein scaffolds of the transport vesicles (ER>Golgi - COPII, Golgi>ER - COPI) (Alb 13-20 (on-line), 13-21 (on-line)). ER exit sites associated with COPII coated vesicular budding represent an integral and indispensable part in the ER resident protein quality control (folding) machinery, i.e. ER associated folding and degradation (ERAF/ERAD).
ER is the first compartment of ESP. Endoplasmic reticulum represents an extensive system of interconnected membrane delimited but communicating membrane bound sheets/stacks. ER continuously extends from nuclear envelope and spreads to cell periphery. In polarized cells, such as neurons, it can be found both in dendrites as well as axons. The membranous system of ER cisternae is functionally polarized. The ribosome-studded ER sheets (rough ER - RER, Alb 12-36 (on-line)) are in continuity with smooth ribosome-free cisternae (smooth ER - SER). These regions of the reticulum (SER) usually function as COPII associated ER exit sites (Alb 12-38 (on-line)). Generally speaking, it seems plausible to assume that ER cisternae contain local intraluminal functional microdomains of folding machinery proteins.
The fraction of SER in the overall ER network is considerably higher in cells with prominent cholesterol turnover, such as those synthesizing steroid hormones (e.g. adrenal cortex), or those responsible for metabolic degradation of toxic or xenobiotic compounds (e.g. therapeutic drugs) through transient hydrophobic chemical intermediates. Hepatocytes are cells typically and extensively involved in such detoxification reactions.
The complex architecture of ER harbors a number of key cellular activities including initial N-linked glycosylation (Alb 12-51 (on-line)) , folding and assembly of proteins, control of Ca2+ signaling and homeostasis, and biosynthesis and successive distribution of lipids (Alb 12-58 (on-line), 12-60 (on-line)). . Protective ER signaling cascades associated with increased amounts of improperly folded proteins may culminate in programmed cell death (see below).
To repeat, proteostasis can be defined as a complex system of pathways involved in cellular and biochemical homeostasis of proteins. These pathways include complex mechanisms of protein synthesis, folding, modification, targeting, as well as degradation of proteins in the cell. It can be stated that ER is fundamentally involved in all these mechanisms. It should be further stressed that ER resident proteostatic molecular machineries work in tight interconnection with ubiquitin-proteasome system (UPS) (see below and Gene Expression) and autophagy pathways (see below) - other two key executor pathways maintaining the dynamic equilibrium of cellular proteomic network.
As mentioned above, proteosynthesis of all cellular proteins starts on free cytosolic ribosomes or polyribosomes (connected by a single molecule of mRNA). The import of the protein into the lumen of endoplasmic reticulum depends on the presence of ER import sequence in the primary amino acid sequence of the newly synthesized (emerging from ribosome complex) protein. Therefore the import of the newly synthesized protein into ER is a co-translational process.
This special N-terminal signal sequence is characterized by a hydrophobic motif, common to leader peptides of all proteins destined for import into ER. These proteins are synthesized on ribosomes associated with endoplasmic reticulum (RER) which thus, on the contrary to SER, constitutes the proteosynthetic "face" of ER.
The co-translational guidance of the emerging protein is mediated by specific binding of the leader peptide /ribosome/mRNA complex to the signal recognition particle/receptor/ trans membrane translocator channel complex in the ER membrane (Alb 12-42 (on-line)). During the ER import, the hydrophobic signal sequence becomes embedded in the ER membrane. The final step in synthesis of luminal (soluble and non-membraneous) proteins is the cleavage of the hydrophobic signal peptide by signal peptidase and release of the nascent protein into the lumen of ER cisternae (Alb 12-46 (on-line))
| Currently, there are at least thirty different inherited monogenic human disorders associated with pathogenic variations in the signal sequences or directly related to aberrant signal sequence processing (for details see Jarjanazi et al., 2008). It can be anticipated that the number of these disorders will increase in time, as it will become evident that such a type of molecular pathology is involved in the genesis of more common maladies (e.g. cancer). The caveats of experimental evaluation of these pathogenic variations originate from their impacts on dynamics of protein synthesis and intracellular protein processing rather than alteration of structural properties of the particular affected (mutated) protein. |
Integral membrane or multiple-transmembrane proteins (Alb 10-17 (on-line)) are also synthesized by the RER associated ribosomes. The membrane insertion of these proteins is a co-translational process that depends on the alternating hydrophobic (transmembrane) and hydrophilic stretches in the amino acid sequence of these molecules. The single or multiple-transmembrane topology is achieved by elaborate use of either co-translational translocation of hydrophilic parts of the protein into ER lumen, anchoring of hydrophobic parts in the environment of the membrane and protein synthesis on the cytosolic face of the ER membrane (Alb 12-47 (on-line), 12-48 (on-line), 12-49 (on-line), 12-50 (on-line)). Integral membrane proteins are retained within the biological membrane until they reach their final cellular destination.
Similarly, GPI anchored proteins are also generated in the ER cisternae(Alb 12-57 (on-line)).
Proteins are synthesized in an unfolded state (Alb 6-80 (on-line)) and have to become properly folded into their native structures (see above) in order to be further processed in the secretory pathway. The estimate is that one third of human genome protein products are processed through ER, therefore get folded there. Guided by the primary information encoded in the polypeptide backbone of the primary amino acid sequence, proteins synthesized into the lumen of ER cisternae undergo folding process to attain an energetically stable conformation. This means that extensive hydrophobic surfaces are packed within the protein interior and intra molecular interactions such as salt bridges or disulfide bonds are established to gain the folded configuration and to minimize the free energy associated with the newly synthesized protein. The conformation attained in ER is not necessarily the final active conformation and may get further modified in successive processing compartments - typically in Golgi apparatus.
Importantly, the kinetic and posttranslational thermodynamic stability of proteins is dependant on the chemical milieu of the ER environment. Once again, there is a difference between luminal and membrane proteins (transmebrane proteins in special). Water soluble proteins are folded within the luminal environment of ER cisternae; membrane proteins, on the contrary, are folded in three different chemical environments (ER lumen, membrane lipid bilayer, and cytosol), which are fundamentally different in terms of pH or Ca2+ concentration.
Folding of a nascent protein in the ER is not a self-catalyzing process, meaning that there are numerous and various ER resident proteins (chaperones) that assist the synthesized proteins to attain their proper native conformations. Similarly to cytosolic proteins (see above) ER folding starts once the protein enters ER lumen and continues within ER till it exits to Golgi (properly folded) or UPS (misfolded).
Folding is principally a process of free energy minimization, and as such may inherently include transient energy barriers that, when simplified, are reduced by action of chaperone molecules (see above). Other general principle of chaperone-mediated folding in ER is its cyclic character, i.e. repetitive rounds of folded protein/chaperone interactions to reach and control the proper conformation (Alb 6-82 (on-line)). Detailed description of the complexity and number of ER resident chaperone proteins and pathways exceeds the extent and purpose of this text. Just to give an example, we would like to mention the cyclic calnexin/calreticulin chaperone pathway that utilizes ER resident N-linked glycosylation of newly synthesized proteins (Alb 12-54 (on-line), Alb 12-51 (on-line)).
Once the proper conformation is reached, the protein can be further processed down the secretory pathway through ER exit sites into COPII coated vesicles and transported via pre-Golgi intermediates to cis-Golgi for additional posttranslational modifications.
The overall efficiency of protein translation in ER has not been exactly set; nevertheless the estimate is that about 30% of newly translated proteins are not correctly synthesized. Similarly, ERAF is also not flawless, and a number of polypeptides in ER do not reach acceptable folding state (conformer). To prevent any potential harmful effects of misfolded proteins (such as intracellular aggregation) ER harbors an elaborate system of elimination of improperly folded proteins. Energetically unstable molecules (misfolded/non-native proteins) are exported out of ER into cytosol through special membrane translocators (Sec61 complex). These aberrant misfolded protein molecules are further modified in the cytosol by covalent linking of polyubiquitin chains. Ubiquitin is a polypetide used for posttranslational protein modifications (see Gene Expression). Ubiquitylation of proteins may be either mono- ubiquitylation (signaling purposes) or poly-ubiquitylation (destined for degradation). Poly-ubiquitylated proteins are targeted to proteasome - cytosolic protein supercomplex capable or efficient protein degradation (Alb 12-55 (on-line), 6-86 (on-line)).
Alternatively to Sec61 complex, a vesicular trafficking system of delivery of misfolded ER luminal proteins to proteasome was recently described. These membranous buds and vesicles originating from ER are not covered by COPII coats, but contain EDEM molecule (endoplasmic reticulum degradation-enhancing α-mannosidase like).
In parallel to ubiquitin-proteasome system (UPS), there is also a continuous constitutive low-level macroautophagy (see below) which complements UPS function in misfolded protein degradation.
Despite the highly effective ER protein degradation system, the amount of misfolded proteins in ER may reach under certain conditions an amount exceeding ERAD capacity. In such a situation, an evolutionary conserved pathway of unfolded protein response (UPR) can be induced in the cell - (Alb 12-56 (on-line)) and may, in its extreme, result in programmed cell death (apoptosis).
| Cellular proteostatic network is a very complex system that can be perceived from different viewpoints (biological, protein biochemical or physical). The basic biological concept: "proteins must be folded properly otherwise they represent a problem for a cell", is a key to proper understanding of the pathogenetic cascades in numerous human diseases, whose common denominator is protein misfolding (Alb 6-85 (on-line), 6-89 (on-line)). These diseases may be caused either by mutations that result in improperly folded proteins, which are not capable of normal function and are preferentially degraded by ERAD system. Or can be due to mutations resulting in aggregation prone protein variants not efficiently degradable by ERAD. Other pathogenic possibilities are mutations compromising whatever constituent of proteostasis network (ERAD, ERAF, UPS or macroautophagy). But most commonly it is relative ERAD insufficiency (e.g. age-related) that results in a continuous low-level toxic effect of misfolded protein molecules. Proteostatic network has lately become a very promising pharmacological target and it can be expected that a number of efficient drugs for previously untreatable protein folding disorders will appear in near future. By regulated modulation of proteostatic processes, these compounds may help the compromised misfolded protein to become functional or, in an opposite fashion, diminish the effects of toxic misfolded species. For additional details about proteostasis, protein folding, protein folding diseases and the perspectives of their treatment see excellent reviews by Aridor, 2007 and Roth et al., 2008. |
Golgi apparatus is the next successive compartment of the early secretory pathway (ESP) on the way of the ER-synthesized proteins destined for exocytosis, cytoplasmic membrane insertion or for lysosomal/endosomal compartment.
Golgi apparatus can be structurally defined as a system of closed membranous stacks/cisterns, which has an entry (cis) and exit (trans) faces with medial cisterns in between. The internal organization of Golgi cisterns may undergo dynamic functional and structural changes in relation to secreted cargo load. There are data demonstrating inter-cisternal communications, partial fusions, and above all, trans-cisternal tubular shortcuts.
cis-Golgi communicates via vesicular tubular structures (pre-Golgi) intermediates with ER exit sites (viz. výše, Alb 13-21 (on-line)). As repeatedly mentioned, only properly folded proteins can exit ER in COPII coated vesicles and enter Golgi apparatus. The transport between ER and Golgi is bi-directional - number of ER resident molecules gets recycled back to ER in COPI coated vesicles on the basis of specific ER retention signals in their sequences.
trans-Golgi is the exit site of Golgi apparatus and assures (by vesicular transport) the delivery of individual protein molecules to their final destination sites: cytoplasmic membrane (exocytosis) and endosomal/lysosomal system.
The membranous flow between ER and Golgi apparatus and successive compartments of the secretory pathway is a part of pan-cellular dynamic equilibrium of membrane trafficking.
Similarly to endosomal system (see below), there is an inherent, functionally important declining pH gradient from ER towards trans-Golgi exit face.
The major functions of Golgi apparatus, within ESP, are posttranslational modifications of proteins by glycosylation. The proteins are initially N-glycosylated on asparagine residues in ER (viz.výše, Alb 12-51 (on-line)), in Golgi apparatus these oligosacharide side chains are further extended to either complex or high-mannose oligosaccharides (Alb 13-25 (on-line), see below in endosomal/lysosomal system). The glycosylation of proteins occurs sequentially by action of successively acting trimming glucosidases or glycosyltransferases that add further carbohydrate moieties. The resultant carbohydrate fraction in the overall protein's molecular weight can be considerable. Proteins (typically proteoglycans) can also be modified by O-linked glycosylation via -OH groups of serines and threonines.
As mentioned above, glycosylation is a sequential process, which is, besides others, achieved by internal functional stratification of Golgi apparatus. It has been proposed that individual enzymatic reactions are enclosed in individual Golgi cisterns and the substrate glycoproteins are processed as they pass through the system of cisterns from cis- to trans-Golgi (Alb 13-26 (on-line), Alb 13-29 (on-line)). There are currently two models of this functional and structural organization of Golgi apparatus: vesicular transport model, which suggests stable enzymatic content of individual Golgi cisterns and vesicular shuttling of substrate glycoproteins in between the cisterns. On the contrary, cisternal maturation model proposes continuous passage of substrate molecules and inter-cisternal transfer of resident enzymatic machinery (Alb 13-30 (on-line)). Recent experimental results prefer, the latter model - cisternal maturation.
To summarize, Golgi apparatus represents a dynamic membranous system of maturing cisterns that assures successive enzymatic modifications of cargo molecules (proteins). It is expected that the load of cargo passing from ER through pre-Golgi intermediates may induce structural changes in Golgi apparatus and is processed as a cargo wave.
The extent of glycosylation of proteins is immense as well as its importance, despite not fully understood yet. Oligosaccharides bound to proteins are build-up from a set of monosacharides despite the absence of any basic information code for the resultant sequence. Glycosylation of newly synthesized proteins is expected to play crucial roles in their folding as well as in their final physical-chemical properties. Oligosaccharides are expected to fundamentally contribute to protein-protein interactions or their antigenic capacity, as the rigid backbones of oligosaccharides tend to protrude from polypeptide chains. For additional details about this topic refer to Biochemistry or Immunology lectures and courses..
| Glycosylation patterns are species specific, a fact important for biotechnological pharmacoproduction. Recombinant proteins, even though with human primary amino acid sequence, acquire glycosylation profiles of the host species, be it either direct production in animals or in non-human cell lines. |
Exocytosis represents a continuous dynamic transport of biological material to the cellular exterior and/or to cytoplasmic membrane. Similarly to endocytosis, exocytosis is an inherent property of every eukaryotic cell. Rates of both these processes as well as the remainder of cellular membrane traffick must remain in a dynamic equilibrium, in order to maintain the constant size of the cellular surface and volume by continuous flow of biological membranes.
Posttranslationally processed water soluble proteins (see above) destined for extracellular space or membrane proteins for cytoplasmic membrane insertion are targeted to nascent secretory granules at the exit sites of Golgi apparatus (trans-Golgi). Limiting membrane of these granules contains hydrophobic proteins such as integral membrane proteins, membrane associated proteins (see above) or glycolipids. These hydrophobic compounds are destined either for cytoplasmic membrane insertion or regulate normal physiological functions of the granules themselves. On the contrary, soluble compounds represent the luminal cargo.
Secretory granules undergo a complex maturation prior their arrival to the sites of exocytosis and release of their contents at the cytoplasmic membrane (Alb 13-56 (on-line)). These may include shedding and recycling of some of the membrane associated coating proteins back to the donor compartment (trans-Golgi) or additional posttranslational modification of the cargo molecules (e.g. proteolytic cleavage or condensation of the contents). The topology of the sites of exocytosis may correlate with functional polarization of the cellular surfaces (e.g. polarized epithelia or neurons) (Alb 13-61 (on-line)). Therefore the docking, membrane fusion and release of the contents of secretory granules can also be polarized (Alb 13-62 (on-line), 13-64 (on-line)).
According to the currently accepted paradigm, exocytosis (secretion) can be functionally sub-classified into two principal types - constitutive (unregulated and permanent) and regulated (Alb 13-54 (on-line), 13-55 (on-line)). The release of the granules follows the general molecular concept introduced above (tethering/docking, membrane fusion, and release of the contents/insertion of membrane proteins) (Alb 13-57 (on-line)).
The process of constitutive (unregulated) secretion is a ubiquitous property of all eukaryotic cell types. The size of the transport vesicles destined for this type of release is within the resolution limit of an electron microscope. Examples of proteins released into the cellular exterior by constitutive exocytosis are plasma proteins secreted by hepatocytes; immunoglobulins by plasma cells; extracellular matrix protein components by fibroblasts, osteoblasts or chondrocytes. Similarly, lysosomal enzymes can be released by osteoclasts or other cell types belonging to monocyte/macrophage cell lineage (images B_cell, mucin_hepatocyte_fibroblast).
Regulated secretion refers (e.g. endocrine or exocrine) to a process, when preformed mature granules are retained in the cytoplasm and are released after a specific stimulus (nervous, chemical or endocrine). Classical secretory granules (e.g. various exocrine and endocrine granules; cytotoxic granules of T lymphocytes; granules of heparinocytes, granulocytes or platelets) are well discernible by optical microscopy (images Ag granules, zymogenic granules).
Endosomal/lysosomal system (ELS) is closely associated with vesicular transport as well as early secretory pathway (ESP). This system represents one of the two (UPS (Alb 12-55) is the other one, see above) main degradation centers of the cell. The whole ELS is extremely dynamic, and is composed of communicating membrane bound vesicles involved in vectorial and regulated cargo transport. In addition, ELS is vastly inducible in terms of its overall volume. Functional and spatial stratification of endosomal - lysosomal compartment is very complex. The tendency to subdivide ELS, whose inherent function is continuous multidirectional flow of membranous constituents and luminal cargo molecules, may result in unwanted simplifications. Generally accepted concept of distinct vesicular populations (early, recycling, late endosomes and lysosomes, see below) with their specified molecular markers should be considered cautiously. The intimate functional connection between late endosomes (mannose-6-phosphate receptor - M-6-PR positive) and lysosomes (M-6-PR negative) resulted in compound designation: late endosomes/lysosomes (see below).
Lysosomes (image lysosomes_3D) are responsible for controlled intracellular digestion of macromolecules and contain approximately 60 - 100 luminal hydrolytic enzymes, comprising proteases, nucleases, glycosidases, lipases, phospholipases, phosphatases and sulfatases. Functional optimum of all these enzymes is at acid pH, which is maintained by vacuolar-type H+-ATPase and chloride protein channel in the lysosomal membrane (Alb 13-31 (on-line)). The proteome (set of all proteins including splicing variants) of lysosomal membranes includes 130-150 different proteins. The most abundant lysosomal membrane proteins are highly glycosylated lysosomal associated membrane proteins (LAMPs) and lysosomal integral membrane proteins (LIMPs). Their high level of glycosylation (with high fraction of degradation resistant polylactosamines) may serve, besides others, as protective barrier against luminal lysosomal proteases.
As stated above, ELS is a continuously and structurally remodeling compartment with widespread contacts in the cell. Spatial coordination of shuttling of membrane and luminal contents is vast, and in majority of aspects not fully understood. The proper cellular targeting and homeostasis of either endocytosed or redistributed material (protein, carbohydrate or lipid compounds) is essential for proper cellular homeostasis. Many of these processes require complex molecular interactions of protein-protein or protein-lipid (membrane) phases.
Biology of ELS will be explained through its biogenesis - coordinated delivery of substrates and structural components necessary for the functional integrity of ELS.
There are three major pathways for entry of substrates into late endosomes/lysosomes - endocytosis, autophagy and phagocytosis. Their individual utilization is cell type specific (Alb 13-35 (on-line)).
Endocytosis is a general term used for the process of internalization of extracellular fluid or material by invagination of cytoplasmic membrane and subsequent intracellular sorting, distribution and recycling. Similarly to exocytosis, every eukaryotic cell is capable of constitutive endocytosis. In addition, specific cell types are capable of regulated, in special cases, receptor mediated endocytosis. Molecular background of endocytosis follows the basic concepts of vesicular transport (see above, Alb 13-41 (on-line)): membrane bending by coating proteins or lipid bi-layer reorganization, release of the vesicles, vectorial transport to the acceptor compartment and parallel recycling of the specific molecules (e.g. receptors or coat proteins) (Alb 13-46 (on-line)). The endocytic process is characterized by two essential prerequisites - molecular sorting (early and late endosomes) and progressive intraluminal pH decrease towards the lysosomes (Alb 13-49 (on-line), 13-50 (on-line)).
Endocytosed molecules are delivered in coated vesicles to small intracellular organelles - early endosomes, which are slightly acid (intraluminal pH 6.0 - 6.2). Endocytosed ligand - receptor complexes dissociate in the pH sensitive manner in early endosomes and the receptors are recycled to the plasma membrane in recycling endosomes. From early endosomes, the substrates pass to late endosomes, where they merge with lysosomal hydrolases to form a single membrane bound compartment - late endosomes/lysosomes. Lysosomes are thought to be produced by maturation process from late endosomes and their pH is about 4.5 - 5.5. The low pH is critical for optimal enzymatic function of luminal hydrolases (see above). The endosomal pathway functions (most probably) on the basis of the "kiss-and-run" principle i.e. the sorting compartments (early and late endosomes) are stationary and the cargo is shuttled between them in transport vesicles.
| It should be stressed that the current knowledge of molecular background of endocytosis is extremely complex and every attempt to lecture it, results in simplifications. Its fundamental property is a constant remodeling through intercompartmental contacts, therefore static description of individual endocytic steps (early endosomes, multivesicular bodies, late endosomes, lysosomes) should be perceived cautiously. As a brief introduction into the complexity of the system see reviews by Johannes and Lamaze, 2002 or Ungewickell and Hinrichsen, 2007. To demonstrate different dynamic aspects of protein sorting refer to roles of ubiquitin protein modifications in endocytic cargo sorting as reviewed by Mukhopadhyay and Riezman, 2007. |
Transcytosis is a variant of endocytosis which can be defined as vesicle mediated transport through cell. Biological material is endocytosed and selectively sorted in the early endosome at one pole of the cell, and is further directed to the opposite pole of the cell where it gets emptied into the extracellular space in an unmodified form (Alb 13-51 (on-line)).
| Some of the most transcytotically active human cell types are endothelial cells and cells forming placental syncytiotrophoblast. The extent of transplacental transfer of nutrients and various protein molecules is immense (typically immunoglobulins). In addition, transcytosis may represent a way of transplacental entry of certain types of infectious (e.g. viruses) or pharmacological agents. For detailed review on transplacental transcytosis see Fuchs and Ellinger, 2004. |
Autophagy as well as endocytosis occurs in all cell types. It represents a complex system of signaling and executive pathways that provide the cell with disposal mechanism of intrinsic contents (proteins, membranes, organelles). Autophagy occurs as a constitutive (continuous) process or can be activated by various stress conditions such as cellular starvation or partial cytoplasmic damage. There are currently three recognized molecular variants of autophagy: macroautophagy/lysosomal system (MALS), microautophagy and chaperone - mediated autophagy (CMA).
Macroautophagy (MALS) designates a process, when an impaired organelle(s) or portion of cytoplasm is delimited by autophagosomal membrane. The exact origin of autophagosomal membrane (de novo synthesis vs. derivate of pre-existing membranes such as cisternae of smooth endoplasmic reticulum) still remains unresolved. Nevertheless the autophagosomal proteome has been defined in mammalian cells. Autophagosomes fuse with late endosomes (see above) to form hybrid organelle autophagolysosome, in which the degradation of the autophagosomal contents by lysosomal hydrolases starts. The hydrolytic products are recycled and reutilized by the cell through a combination of passive diffusion, specific transmembrane transporters or membrane contacts.
Microautophagy is a process, in which the lysosomal membrane directly engulfs a part of the cytoplasm or small organelles such as peroxisomes (pexophagy).
Chaperone - mediated autophagy is, on the contrary to MALS, a highly selective protein degradation mechanism. It requires a specific targeting sequence signal (KFERQ-like motifs) in the primary amino acid sequence on the substrate proteins. Molecular chaperones in the cytosol and in the lysosomal lumen (hsc70 and ly-hsc70) and lysosomal membrane receptor LAMP2a (isoform of LAMP2 protein) are essential for the direct transport of substrate proteins across the lysosomal membrane. CMA is activated under stress conditions, such as nutrient deprivation, oxidative stress and exposure to toxic compounds.
| Autophagy is an evolutionary conserved self-protective cellular pathway, which is an integral part of the cellular proteostatic regulatory network together with the UPS (see above). Physiological roles of autophagy as well as its implications for human pathology are vast and include participation in control of cell death programs (for detailed review see Bredesen, 2007), physiological cellular protein clearance or even normal human ontogenesis and aging (see Terman et al., 2007). As an example, autophagy seems to be one of the major glycogen mobilizing pathways in newborns. The amount of data about autophagy has virtually exploded during the last few years. It can be anticipated that the fundamental roles of autophagy malfunction have already been or will be demonstrated in many major human pathologies including neurodegenerative disorders or cancer (see reviews by Cuervo, 2004, 2006). |
Phagocytosis is the last major entry pathway into lysosomes. It is utilized for degradation of extracellular material such as microorganisms and dead cells after their ingestion by cytoplasmic membrane (Alb 13-39 (on-line), 13-50 (on-line-)). Phagocytosis is a process restricted to specialized cells of immune system such as macrophages, neutrophil granulocytes or dendritic cells. The phagosomes fuse with late endosomes and the ingested material is then degraded in late endosomes/lysosomes.
| Additional details about this very important aspect of cellular biology will be given in specialized Microbiology and Immunology courses. Nevertheless, it should be kept in mind that the fundamental concepts of phagocytic function and related immunobiological processes (e.g. antigen processing and presentation) are derived from cellular biology of cytoplasmic membrane and ELS. |
Lysosomal hydrolases and membrane proteins are synthesized into the cisternae of rough endoplasmic reticulum, fold there and exit ER through COPII vesicles. They further pass through the Golgi apparatus to the trans-Golgi network. The transport vesicles arise by budding from the trans-Golgi and deliver these proteins to late endosomes, which serve as molecular sorters in the endosomal-lysosomal system (see above).
Most of the soluble lysosomal enzymes are posttranslationally modified with N-linked high mannose-type oligosaccharides (Alb 13-25 (on-line))(see above). The most important modification for targeting and sorting of lysosomal proteins is the formation of the mannose-6-phosphate (M-6-P) recognition marker. The M-6-P recognition signal is generated in a two step reaction. In the first step, the lysosomal enzymes are phosphorylated by UDP(uridine diphosphate)-N-acetylglucosamine-1-phosphotransferase (phosphotransferase). This enzyme transfers N-acetylglucosamine-1-phosphate from UDP-N-acetylglucosamine to the C6-hydroxyl group of a selected mannose residue on the high mannose-type oligosaccharide of the particular lysosomal protein. In the following step, the N-acetylglucosamine residues are cleaved by the enzyme N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (phosphoglycosidase) (Alb 13-38 (on-line)). This second reaction exposes the M-6-P recognition marker and allows the lysosomal enzyme to be recognized by mannose-6-phosphate receptors (M-6-PRs) localized in the membranes of trans-Golgi. Because most lysosomal hydrolases contain multiple oligosaccharides, they may acquire more than one M-6-P marker residue, which thus provides a higher affinity signal for M-6-PRs.
The receptor proteins (M-6-PRs) bind lysosomal hydrolases by their luminal domains oriented into trans-Golgi cisterns. The trans-Golgi derived vesicles deliver their contents to late endosomes, where the low pH induces dissociation of lysosomal enzymes from M-6-PRs. The hydrolases are afterwards dephosphorylated and M-6-PRs are recycled back to trans-Golgi or alternatively to cytoplasmic membrane (Alb 13-37 (on-line)). Some of the newly synthesized lysosomal enzymes escape the binding to M-6-PR in the trans-Golgi and may get exocytosed. MPRs localized at the plasma membrane are capable of binding the escaped M-6-P-containing lysosomal enzymes and returning these molecules by receptor - mediated endocytosis to lysosomes via early and late endosomes.
Some lysosomal hydrolases such as cathepsin B, acid α-glucosidase or lysosomal acid phosphatase are delivered to late endosomes/lysosomes in their precursor forms and must be further posttranslationally cleaved by autocatalytic proteolysis or by other lysosomal resident proteases (e.g. cathepsins).
There are other alternative mechanisms of delivery of soluble lysosomal enzymes to late endosomes/lysosomes besides the M-6-PR-mediated trafficking. Prosaposin, a precursor of four lysosomal saposins (A - D), acid sphingomyelinase, GM2 activator protein, and cathepsins D and H share a common intracellular receptor - sortilin - for sorting from trans-Golgi to late endosomes/lysosomes. Beta-glucocerebrosidase employs LIMP2 as a receptor molecule in the lysosomal membrane. Acid phosphatase most probably utilizes mannose-phosphate mediated recycling pathway to lysosomes from cytoplasmic membrane.
The lysosomal membrane proteins (LMPs), such as LAMPs, LIMPs and other, are sorted from trans-Golgi to late endosomes/lysosomes by M-6-P independent mechanism. These proteins leave trans-Golgi in clathrin coated vesicles distinct from those that transport the M-6-P - tagged hydrolases and get delivered to late endosomes/lysosomes on the basis of either tyrosine or di-leucine targeting signals in their cytoplasmic C-terminal tails.
| Lysosomal storage disorders (LSDs) represent a group of ~50 monogenic human diseases; their overall incidence is 1:6000 newborns. Common denominator shared by all these diseases is the expansion of lysosomal compartment in affected cell types by improperly processed substrates, resulting in foamy transformation of the cytoplasm. Majority of these disorders is caused by pathogenic genetic variations in individual lysosomal hydrolases, their activators or are due to improper posttranslational lysosomal protein processing. These disorders are lysosomal enzymopathies. Additional major group of lysosomal storage disorders represents selective malfunctions of various late endosomal/lysosomal membrane proteins. These diseases can be generally described as defects of the endosomal-lysosomal system itself. Understanding the molecular pathology of LSDs was repeatedly made possible through studies of endosomal/lysosomal biology and its systemic cellular interconnections. Based on the fact that the late endodomal/lysosomal proteome includes aproximately 200-250 proteins, it is plausible to assume that the overall number of lysosomally associated diseases (not necessarily accompanied by storage) defined on the genetic level will increase. |
The characteristics of lysosomes (acid pH, presence of luminal acid hydrolases, LAMPs and absence of mannose-6-phosphate receptor - M-6-PR) are shared by a group of cell type specific organelles, which are called lysosome - related organelles (LRO). LROs include melanosomes of melanocytes, lytic granules of T lymphocytes, platelet - dense granules, or basophil and azurophil granules of granulocytes. Common property of all LROs (differentiating them from classical lysosomes) is their exocytic ability. Additionally, LROs contain biologically active substances. The extent of participation of LROs in the intracellular degradative pathways is minimal, despite the content of lysosomal hydrolases.
| Common pathology of lysosomes and LROs represents cellular basis of diseases such as Chediak-Higashi, Heřmanský-Pudlák or Griscelli syndromes - diseases associated with immunodeficiency, skin pigmentation abnormalities (albinism) and hemorrhagic diathesis (bleeding). All these symptoms are more or less associated with impaired exocytosis of LROs. |
Mitochondria represent a membranous cellular compartment that participates in a number of intermediary metabolic pathways - energy production being the major one. Approximately 95% of cellular ATP is generated by oxidative phosphorylation (OXPHOS), a metabolic pathway residing in mitochondria (Alb 14-10 (on-line)). Other mitochondrial metabolic pathways such as citric acid cycle, beta oxidation of fatty acids, parts of urea cycle and porphyrin synthesis will be mentioned during other courses (e.g. biochemistry) in detail.
The evolutionary endosymbiotic bacterial origin makes mitochondria rather unique compartment in eukaryotic cells (Alb 14-70 (on-line)). Nevertheless, similarly to other membranous organelles, mitochondria are of maternal origin and are a derivate of pre-existing structural substrate, i.e. cannot be synthesized de novo (see above).
Structural feature differentiating mitochondria from other organelles is their doubled biological membrane. This setup results in two structurally and functionally distinct mitochondrial sub-compartments (inter- membrane space and mitochondrial matrix) delimited by outer and inner mitochondrial membranes, and various intra-mitochondrial trans-membrane gradients (Alb 14-8 (on-line)).
Outer mitochondrial membrane has a plain shape without any excessive membrane invaginations and is connected by a number of contact spots with the inner mitochondrial membrane. Some of the protein components of the outer mitochondrial membrane function as receptors for binding and transmembrane transport of mitochondrial proteins (coded by gDNA), which are synthesized on the cytoplasmic ribosomes.
Inner mitochondrial membrane has a considerably more complex invaginated structure - it forms a number of variously shaped (mostly flattened stacks) cristae. Inner mitochondrial membrane delimits the space occupied by mitochondrial matrix, which harbors numerous metabolic pathways. Multisubunit transmembrane protein complexes (4 respiratory chain complexes and ATP synthase (ATPase) complex (Alb 14-15 (on-line)) of the oxidative phosphorylation apparatus (OXPHOS) are also embedded in the inner mitochondrial membrane. OXPHOS system complexes spatially cluster within the inner mitochondrial membrane into protein supercomplexes called respirasomes.
Mitochondria have a semiautonomous genome of maternal origin, formed by circular molecule of DNA (mitochondrial DNA - mtDNA). One mitochondrion contains, on average, 10-20 individual mtDNA molecules, which are organized into nucleoprotein complexes (nucleoids). It is assumed that a single mtDNA nucleoid contains approximately 6 mtDNA molecules. These nucleoprotein complexes are coordinately anchored to inner mitochondrial membrane and also possibly to outer mitochondrial membrane via additional specialized membrane proteins (demonstrated by studies in yeast). Additional interaction of mtDNA nucleoids with cytoplasmic actin cytoskeleton through both mitochondrial membranes has also been proposed. The distribution of mtDNA nucleoids within the mitochondrial network is uneven, resulting in local accumulations as well as regions of the network relatively devoid of mtDNA (for additional details see below). Summarized, the overall number mtDNA molecules in a single cell (!cell type dependant and variable!) is 10-20 000 (1000 mitochondria/cell, 10-20 mtDNA molecules/mitochondrion).
Mitochondrial DNA codes 13 subunits of different complexes of OXPHOS system, 12S rRNA, 16S rRNA and 22 tRNAs, therefore mitochondria are capable of intrinsic transcription and translation as their genome codes for essential molecular constituents of these processes. Nevertheless, it is obvious that the overwhelming majority of mitochondrial proteins (1500 predicted and 900-1000 confirmed up to date) are coded by the nuclear gDNA and these proteins must be targeted and imported to mitochondria from cytosol (see below, Alb 14-51 (on-line), 14-64 (on-line)).
The difference between nuclear and mitochondrial genetics is fundamental. Nuclear genome is diploid (half paternal/half maternal) and becomes transiently (in S-phase) tetraploid to become after mitosis diploid in the daughter cells. Any deviation in the number of chromosomes (aneuploidy) may have considerable pathogenic consequences. On the contrary, mitochondrial DNA is of maternal origin (any paternal leakage of mtDNA - is either efficiently eliminated or massively diluted in maternal cytoplasm of the zygote). The number of mtDNA molecules in a single cell is high (see above), i.e. eukaryotic cells are polyploid for mtDNA. Additionally, the overall amount of mtDNA may physiologically vary throughout the cell cycle or due to mitochondrial degradation (see below). In comparison to gDNA (synchronous replication in S-phase), mtDNA replicates asynchronously and the distribution into daughter cells during cytokinesis is directly related to mitochondrial network dynamics and local mtDNA content within the mitochondrial network. Even though not fully understood yet, it can be assumed that the nuclear cell cycle (gDNA) is somehow interconnected with mtDNA cycle, as both daughter cells receive approximately similar amounts of mitochondrial mass as the original parent cell.
As mentioned above, mitochondrial proteins coded by nuclear gDNA (overwhelming majority) are synthesized on free cytosolic ribosomes or polyribosomes in their precursor forms. Similarly to other types of proteins, mitochondrial proteins are also generally either water soluble or membranous and must be folded and specifically targeted to proper mitochondrial sub-compartments - inter-membrane space, and mitochondrial matrix or to outer and inner mitochondrial membranes. The signal sequences located in the N-termini assure specific binding of mitochondrial proteins to receptors/membrane translocators in the outer and sequentially inner mitochondrial membrane. Mitochondrial proteins are initially folded in cytosol by cytosolic chaperone systems; their transfer through transmembrane translocators is in unfolded state. The final folding and targeting, assembly of complexes included, occurs inside mitochondria and employs mitochondrial machinery of chaperones, folding and assembly factors. It is a concerted action of proteins like cytosolic and mitochondrial heat shock chaperone proteins 70 (c/mtHS70) and TOM (outer membrane) and TIM (inner membrane) and OXA translocators that guarantee the proper final delivery of each individual mitochondrial protein (Alb 12-24 (on-line), 12-25 (on-line), 12-26 (on-line)). It should be pointed out that mitochondrial proteins are multisubunit protein supercomplexes, which in addition to proper targeting require correct intra-mitochondrial folding and assembly - a mechanism relying on additional group of mitochondrial proteins (assembly factors). There is evidence of a separate intra-mitochondrial proteodegradative system, which assures proper intra-mitochondrial proteostasis.
Mitochondria cannot be (according to the current state of knowledge) synthesized de novo, but are derived from pre-existing structural substrate of other mitochondria. Mitochondrial division occurs after the parent mitochondrion gains the necessary size. This process of mitochondrial "doubling" seems to be distinct from mitochondrial network remodeling by fission (described below). The increase of mitochondrial mass can be enormous under certain non-physiologic conditions. Affected cells (called oncocytes) may have their cytoplasm virtually packed with mitochondria (image oncocytes_cirrhosis). This type of cellular transformation can be observed in a number of primary mitochondrial disorders (see below) (image MELAS_muscle) or in some tumors - oncocytic carcinomas.
Degradation of individual mitochondria or parts of mitochondrial network is exerted through macroautophagy in lysosomes (MALS, see above).
Mitochondria are also essential for execution of caspase mediated apoptosis (programmed cell death) mediated by the release of cytochrome C into cytosol.
Mitochondria function as a complex three dimensional network which is continuously remodeled by fusion and fission events (Alb 14-53 (on-line), 14-54 (on-line)). Ratio of these events must remain in a dynamic equilibrium; otherwise the cell suffers from fragmented or elongated mitochondria. Number of proteins and molecular pathways is indispensable for regulation of these fusion/fission dynamics. Complex mitochondrial network remodeling is absolutely essential for many fundamental cellular processes such as apoptosis, cytokinesis, or maintainence of the cellular polarity. It should not therefore be surprising that the number of diseases due improper function of some the key protein regulators of mitochondrial network dynamics is growing. In addition, as has been recently demonstrated, mitochondria and peroxisomes (see below) may structurally interact and defects in this previously neglected interaction may also result in human disease phenotype(s).
| Mitochondrial genome is of maternal origin; therefore the inheritance of traits (pathology included) coded by mtDNA is not Mendelian but maternal. Mitochondrial DNA mutations can be inherited (germinal mutations) or occur during the development and are thus somatic. Due to the nature of intra-mitochondrial environment (e.g. high content of reactive oxygen species) number of mtDNA mutations increases throughout postnatal life. Additionally, mitochondria posses a rudimentary and considerably less efficient DNA repair machinery in comparison to cell nucleus (gDNA). Integrity of mitochondrial genome depends on nuclear genome. Global depletions of mtDNA and resultant deficits of corresponding mitochondrial proteins (see above) or multiple mtDNA deletions are caused by mutations in nuclear genes coding regulators of mtDNA turnover and mtDNA quality control machinery. Nuclear genome codes the major fraction of mitochondrial proteins including the majority of respiratory chain proteins and assembly factors (Alb 14-64 (on-line)). The above stated is necessary to understand the molecular background of two basic types of inherited mitochondrial disorders. One group of these maladies is caused by pathogenic variations (mutations) in genes coded by gDNA - these disorders are inherited as Mendelian traits. The second group of diseases is due to mutations in genes coded by mtDNA (13 subunits of respiratory chain complexes or 22 tRNAs). Compared to mutations in nuclear genes, mutations in mtDNA have a critical threshold effect (only a fraction of all mtDNA molecules carries the mutation), starting in the fertilized egg and further propagating in the developing organism. The resultant mosaic of normal and mutated mtDNA molecules in mitochondria in various cell types is a result of successive cytokinetic events, which distribute the mitochondrial network and mitochondrial DNA into daughter cells and thus tissues and organs. The regulation of these processes i.e. what determines the distribution of normal vs. mutated mtDNA remains largely unknown despite intensive research efforts. Respiratory chain is formed by multisubunit (4-40 subunits) protein super complexes. Proper function of these super complexes depends on proper synthesis, cytosolic transport, transmembrane translocation and final assembly mediated by a number of assembly factors. Some mitochondrial disorders are caused by mutations in these gDNA encoded assembly factors. Assembly malfunction results in degradation of otherwise normal subunits and thus deficiency of enzymatic function. It is also the mitochondrially synthesized phospholipid cardiolipin (enriched in the inner mitochondrial membrane), which is indispensable for assembly of several inner mitochondrial membrane protein complexes. For review of mitochondrial biology, disorders, and therapeutic perspectives refer to excelent review by Yamada and Harashima, 2008. |
Peroxisomes are delimited by a single layer of biological membrane enclosing a finely granular matrix. The size of peroxisomes ranges between 0.05 (also called microbodies) and 2 microns. In most of the cases, peroxisomes do not reach the size of mitochondria. Peroxisomes are a ubiquitous organelle present in nearly all cellular types of human body excluding red blood cells and sperm cells. Peroxisomes are most abundant in hepatocytes, renal tubular epithelium and oligodendroglia (cells synthesizing myelin).
Human peroxisomal proteome (membranous and matrix proteins) consists of products of approximately 80 genes coded by nuclear gDNA. Majority (60%) of these proteins are associated with peroxisomal metabolic pathways. The remaining third are called peroxins and play crucial roles in peroxisomal biogenesis and sustenance. The marker (exclusively present) enzyme of peroxisomes is catalase which can be detected either histochemically (by its enzymatic activity) or immunohistochemically (by specific antibody). In addition, there are numerous antibodies binding peroxisomal membrane proteins or other peroxisomal enzymes that can also be used for detection of peroxisomes.
The biogenesis of peroxisomes (growth and fission) is in many aspects an unresolved enigma; especially in terms of the possible de-novo ER related peroxisomal synthesis. Additionally, peroxisomes have been recently demonstrated to share some proteins essential for biogenesis (fission machinery) with mitochondria. Biogenesis can be, to a limited extent, compared to lysosomal biogenesis - synthesis and transport of structural membrane proteins and matrix proteins (predominantly enzymes).
In comparison to mitochondria, peroxisomes do not carry separate genetic information in the form of any type of nucleic acid. The whole peroxisomal proteome (set of all peroxisomal proteins) is coded by nuclear gDNA and majority of translation of peroxisomal proteins (matrix proteins and a fraction of membrane proteins) occurs on free cytosolic ribosomes or polyribosomes. Targeting of these peroxisomal proteins into the pre-existing peroxisomes is mediated by short specific amino acid signal sequences (peroxisomal targeting sequence - PTS). Principally, there are two types of these sequences in various peroxisomal proteins - (i) C-terminal SKL motif and (ii) C-terminal PTS1 and N-terminal PTS2 motifs. On the contrary to ER and mitochondrial proteins (see above), these peroxisomal proteins are imported from the cytosol in the folded state, though the exact molecular mechanism of this transmembrane transfer is still under study.
The rest of peroxisomal membrane proteins are targeted to peroxisomes from ER presumably by vesicular transport. It has also been recently demonstrated that a fraction of peroxisomal proteins reaches the organelle via mitochondria. Transient interactions of peroxisomes with smooth endoplasmic reticulum were also observed. The issue of retrograde transport to ER and mitochondria from peroxisomes remains enigmatic.
The import of proteins results in the volume expansion of peroxisomes, which culminates in peroxisomal fission. Some of the proteins necessary for peroxisomal fission are also essential components of mitochondrial fission machinery.
Peroxisomes are degraded by microautophagy (pexophagy, see above). It seems that certain proteins of peroxisomal membrane are key players and mediators of this process together with the proteins of lysosomal membrane.
Peroxisomes harbor numerous metabolic pathways. Currently, there are approximately 50 different resident enzymes described in peroxisomes. The degradative (catabolic) functions of peroxisomes include various oxidative reactions of numerous substrates (alcohols such as ethanol, xenobiotics or amino acids) mediated by oxidases utilizing molecular oxygen species with parallel genesis of hydrogen peroxide, which is further efficiently metabolized by the above mentioned enzyme catalase. Other peroxisomal resident metabolic pathways include oxidation of long chain fatty acids (C24, C26), dicarboxylic acids, branched-chain fatty acids and prostaglandins. Peroxisomes also harbor catabolism of purines, glycerol synthesis and retinoid metabolism.
Rodent peroxisomes contain additional enzyme - urate oxidase (forming central crystaloid structures), which converts uric acid to soluble allantoin.
| All peroxisomal functions are directly dependant on a continuous peroxisomal biogenesis, i.e. on the import of peroxisomal membrane and matrix components. This feature of peroxisomal biology also fundamentally differentiates peroxisomal disorders from mitochondrial diseases. There are currently ~24 different monogenic disorders associated with peroxisomal dysfunction. These disorders can be classified into two basic groups : single enzyme deficiencies due to primary defect in particular peroxisomal enzyme (in structurally intact peroxisomes) or peroxisome biogenesis disorders - Zellweger syndrome. Zellweger syndrome (peroxisomal abiogenesis) can be given as a good example of the latter type of peroxisomal disorder. Even though all peroxisomal enzymes are properly synthesized, the target compartment is missing. This results in empty peroxisomes, so-called "ghosts". Peroxisomal enzymes, even though fully functional, remain in the cytosol and are consequently degraded. Peroxisomal substrates can not be handled and eventually become toxic for the cell (e.g. the above mentioned very long chain fatty acids). These compounds may integrate into biological membranes and thus destabilize them. One of the extreme outcomes of this process could be demyelinization (leukodystrophy) As noted above, biogenesis of peroxisomes (fission) shares protein machinery with mitochondria. A lately described human inherited disorder characterized by joint defect of mitochondrial and peroxisomal fission due to mutations in one of the dynamin-like fission GTPases (see above) just documents the importance of this biological interconnection. It has also been proposed that some of the more common liver pathologies such as non-alcoholic steatohepatitis result from failure of peroxisomal function. For additional details about peroxisomal biology and pathology refer to Schrader and Fahimi, 2008 |
The central problem of cytokinesis is that the cytoplasmic cleavage has to happen coordinately with the rest of cell cycle - at the right time (after chromosome separation) and at the right place (to ensure that each daughter cell will get one complete set of chromosomes). The fundamental question thus states: where and how to put the cut?
The biological phenomenon of cytokinesis can be perceived from two different angles: (i) the actual separation of two daughter cells by separation of their cytoplasmic membranes and (ii) mechanisms involved in regulated division of cytoplasmic compartments.
Cytokinesis is a complex phenomenon that includes thorough regulation and fundamental architectural remodeling of cytoplasm as well as cytoplasmic membrane. During cytokinesis, the cytoplasm is divided in two by a contractile ring (Alb 18-34 (on-line)) composed of actin and myosin II filaments, which starts to assemble when the activity M-Cdk (see Cell Cycle and Cell signalization) decreases. Contractile ring drives the ingression of the membranous cleavage furrow (Alb 18-35 (on-line)) between the two newly constituted nuclei. After mitosis, the position of the mitotic spindle determines the location of the contractile ring. The contractile ring is formed in the plane of the methaphase plate, perpendicular to the long axis of the mitotic spindle. Prior to pinching the cell into two daughter cells a narrow cytoplasmic bridge connects the two daughter cells. This bridge contains interdigitating bundles of anti-parallel microtubules with central density called midbody (Alb 18-35 (on-line)). The start of cytokinesis is regulated by polo-family kinases, by yet unknown mechanism.
Majority of mitotic divisions are symmetric; each of the daughter cells receives equal amounts of cytoplasmic contents, similarly to equal amounts of nuclear DNA (chromosomes). There are many unresolved, but fundamental questions related to the regulation and mechanisms of this structural cytoplasmic segregation. What happens with the membranous organelles during mitosis? Does the organellar mass double prior the mitosis, or do the organelles have to be re-synthesized to gain pre-mitotic levels in daughter cells? Is the segregation of organelles into daughter cells random? How are the rates of exocytosis and endocytosis and remodeling of cytoplasmic membranes associated with thorough remodeling of cytoplasmic membrane? Unfortunately, conclusive generally applicable answers for majority of these questions have not been found, yet.
Just to give an example of an organellar behavior during mitosis, let us concentrate on two selected membrane delimited compartments - Golgi apparatus and endocytic compartments. In the interphase Golgi apparatus accumulates around the centrosome in a microtubule regulated fashion. It has been demonstrated that membrane stacks of Golgi apparatus and endosomal membranes split into two clusters associated with doubled centrosomes during prophase and successively disintegrate in metaphase. During late mitosis (anaphase and telophase) the reconstitution of Golgi apparatus in daughter cells originates from two newly emerging clusters (smaller and bigger in size), which are in a close spatial relation of the newly formed nuclear envelope and follow the formation of cleavage furrow. It is these two clusters that coordinately fuse to form new Golgi apparatus. Post-Golgi clusters in the vicinity of midbody contribute to remodeling of the cleavage furrow by fusion of their membranes with cytoplasmic membrane. Despite very convincing experimental data, it is not clear how this process occurs in different cell types or in asymmetric mitosis i.e. whether it is universally valid.
Similarly, mitochondrial network (see above) also segregates into daughter cells in a coordinated fashion. In case of this network, one must keep in mind that mitochondrial network contains embedded genetic information (mtDNA). Therefore inheritance of whatever mtDNA encoded trait is directly dependant on the division of the mtDNA immediate structural environment, i.e. mitochondrial network.
To conclude, cytokinesis is a complex and higly regulated process. Cytokinesis is not a random division of cytoplasm and it definitely is not a mere appendix of nuclear division. (For details see review by Barr and Gruneberg, 2007 or Montagnac et al., 2008.)
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002/2007). Molecular Biology of the Cell, 4/5th edn (London, Garland Science).
Aridor, M. (2007). Visiting the ER: the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Advanced drug delivery reviews 59, 759-781.
Barr, F.A., and Gruneberg, U. (2007). Cytokinesis: placing and making the final cut. Cell 131, 847-860.
Bredesen, D.E. (2007). Key note lecture: toward a mechanistic taxonomy for cell death programs. Stroke; a journal of cerebral circulation 38, 652-660.
Cai, H., Reinisch, K., and Ferro-Novick, S. (2007). Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Developmental cell 12, 671-682.
Cuervo, A.M. (2004). Autophagy: in sickness and in health. Trends Cell Biol 14, 70-77.
Cuervo, A.M. (2006). Autophagy in neurons: it is not all about food. Trends Mol Med 12, 461-464.
Fuchs, R., and Ellinger, I. (2004). Endocytic and transcytotic processes in villous syncytiotrophoblast: role in nutrient transport to the human fetus. Traffic (Copenhagen, Denmark) 5, 725-738.
Jarjanazi, H., Savas, S., Pabalan, N., Dennis, J.W., and Ozcelik, H. (2008). Biological implications of SNPs in signal peptide domains of human proteins. Proteins 70, 394-403.
Johannes, L., and Lamaze, C. (2002). Clathrin-dependent or not: is it still the question? Traffic (Copenhagen, Denmark) 3, 443-451.
Montagnac, G., Echard, A., and Chavrier, P. (2008). Endocytic traffic in animal cell cytokinesis. Current opinion in cell biology 20, 454-461.
Mukhopadhyay, D., and Riezman, H. (2007). Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science (New York, NY 315, 201-205.
Roth, J., Yam, G.H., Fan, J., Hirano, K., Gaplovska-Kysela, K., Le Fourn, V., Guhl, B., Santimaria, R., Torossi, T., Ziak, M., et al. (2008). Protein quality control: the who's who, the where's and therapeutic escapes. Histochemistry and cell biology 129, 163-177.
Roy, S., Zhang, B., Lee, V.M., and Trojanowski, J.Q. (2005). Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol (Berl) 109, 5-13.
Schrader, M., and Fahimi, H.D. (2008). The peroxisome: still a mysterious organelle. Histochemistry and cell biology 129, 421-440.
Terman, A., Gustafsson, B., and Brunk, U.T. (2007). Autophagy, organelles and ageing. The Journal of pathology 211, 134-143.
Ungewickell, E.J., and Hinrichsen, L. (2007). Endocytosis: clathrin-mediated membrane budding. Current opinion in cell biology 19, 417-425.
Yamada, Y., and Harashima, H. (2008). Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Advanced drug delivery reviews 60, 1439-1462.
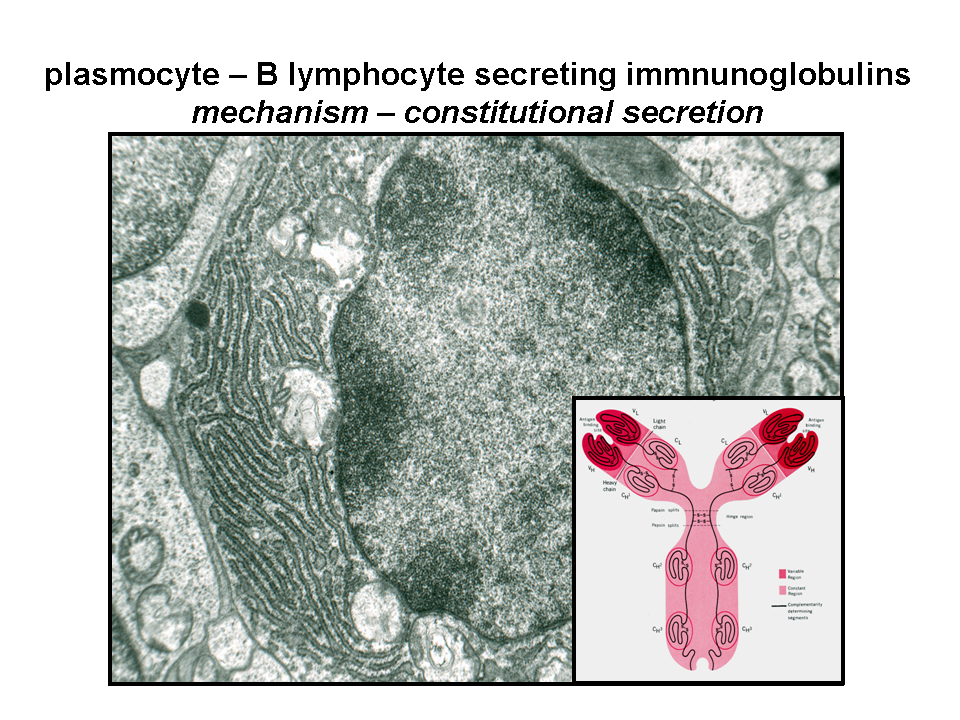
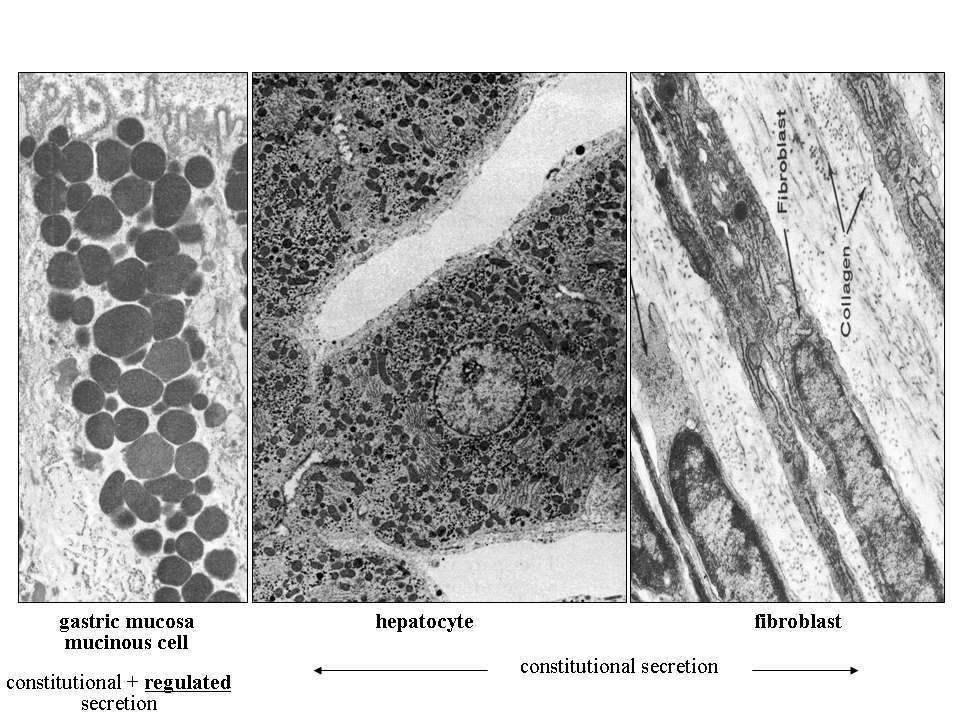
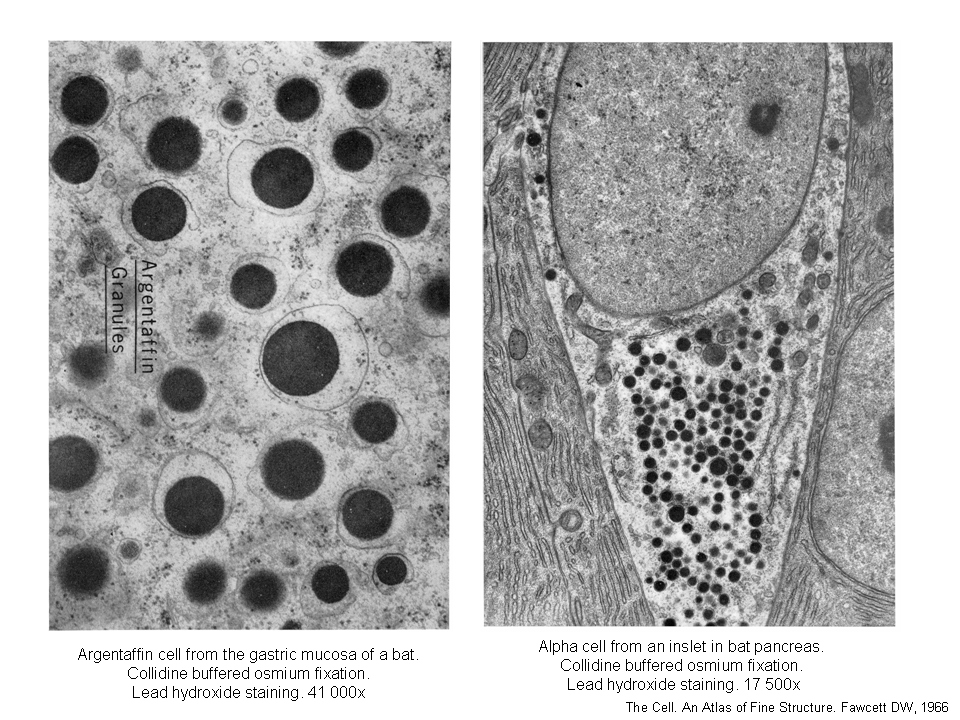
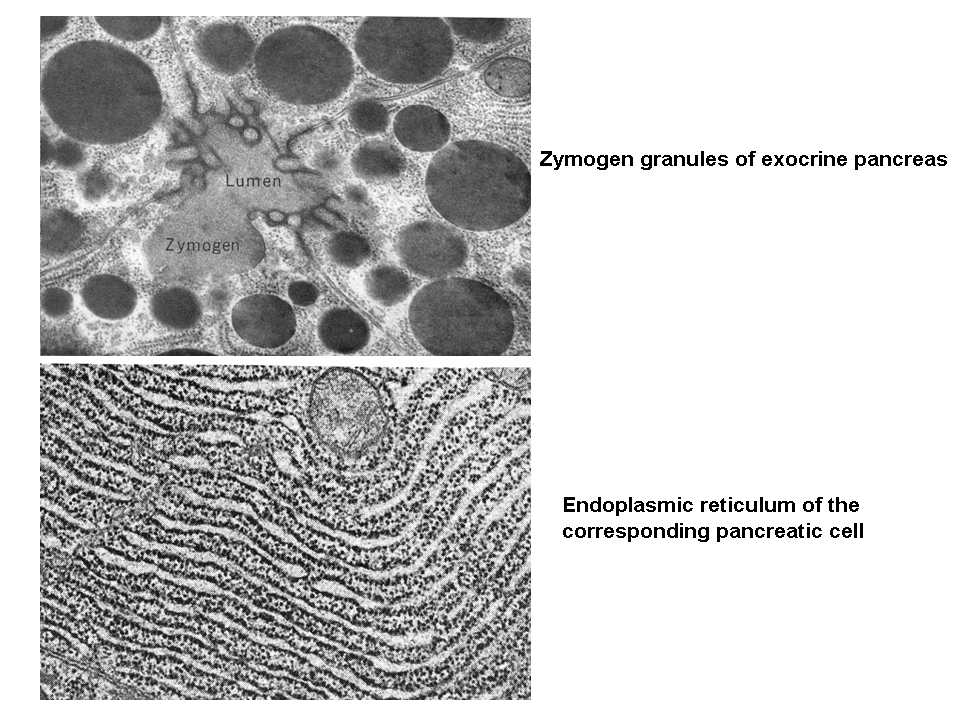
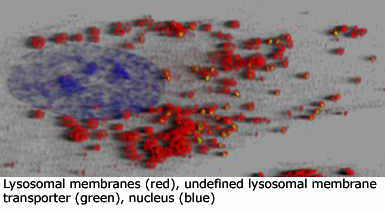
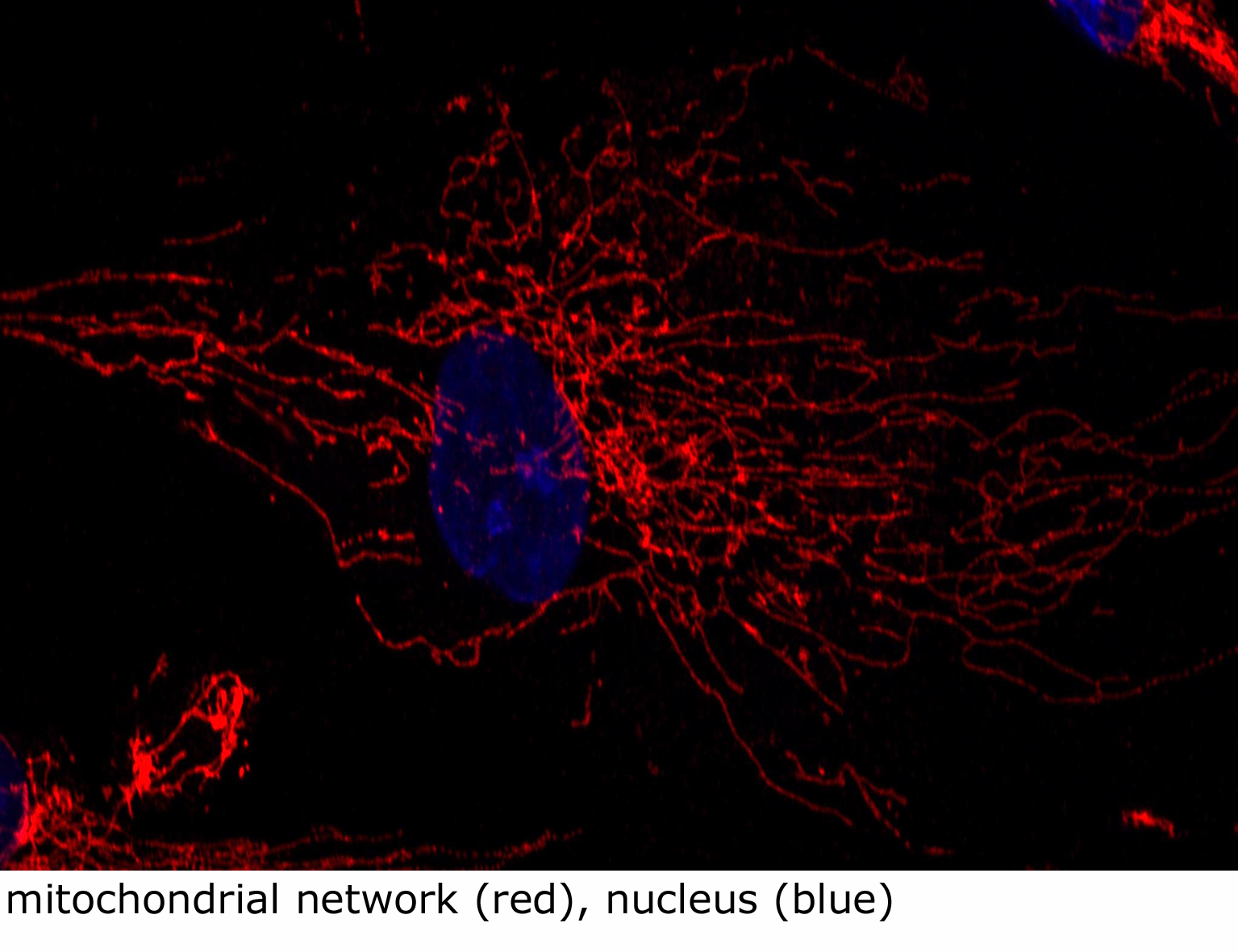
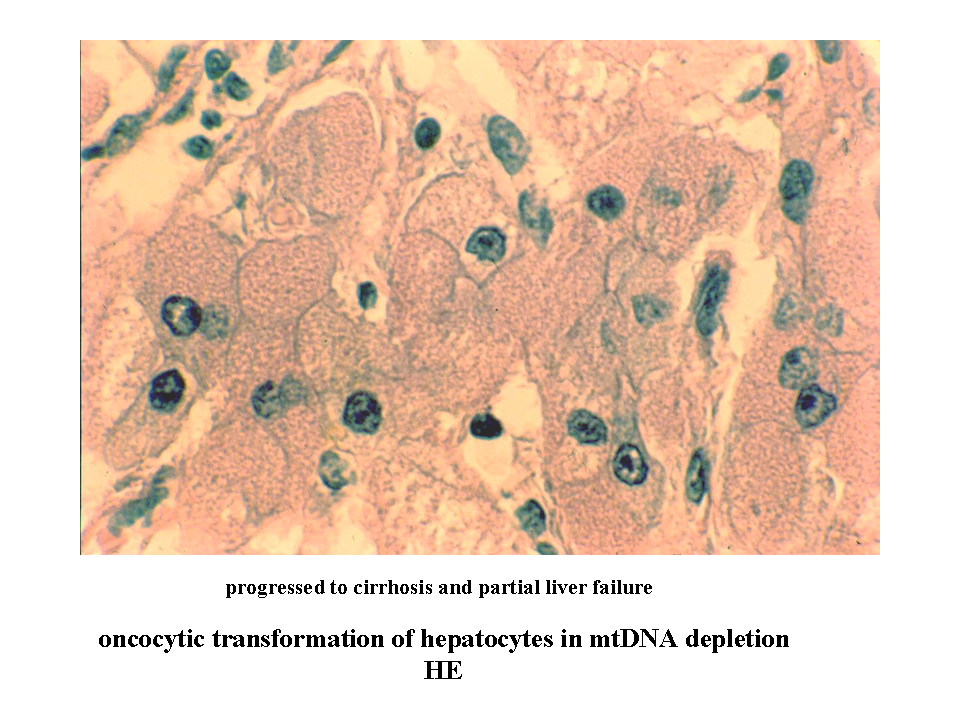
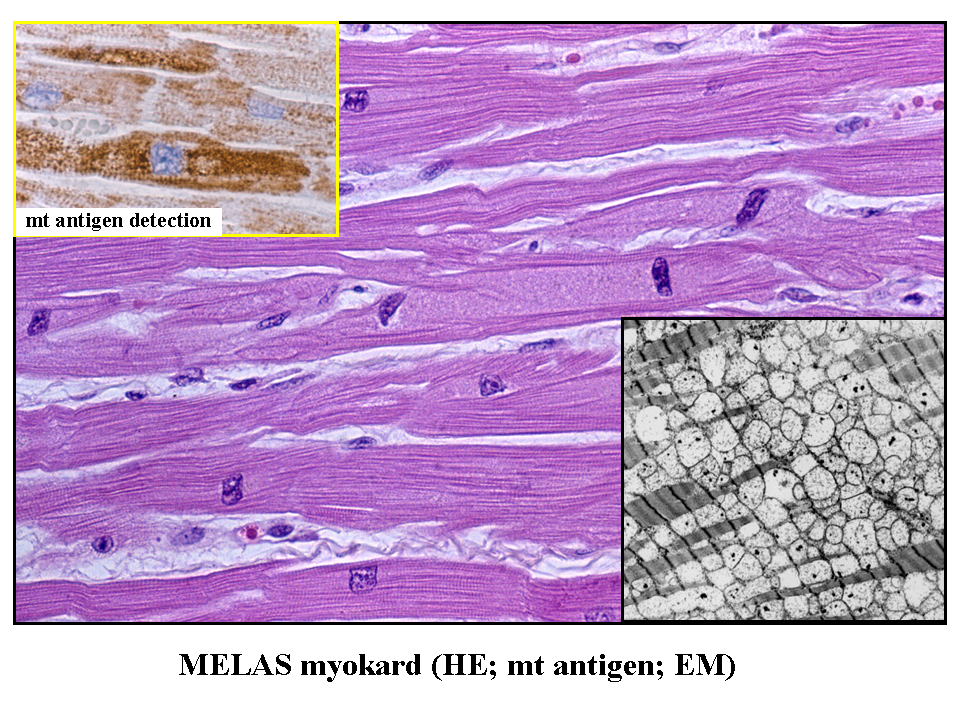
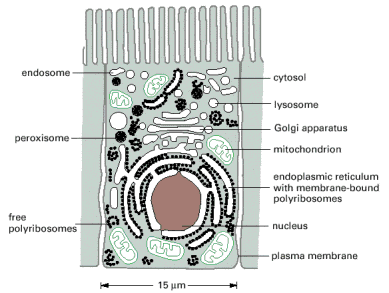{kind=link}
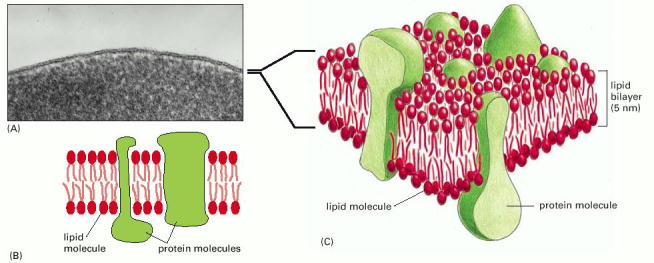{kind=link}
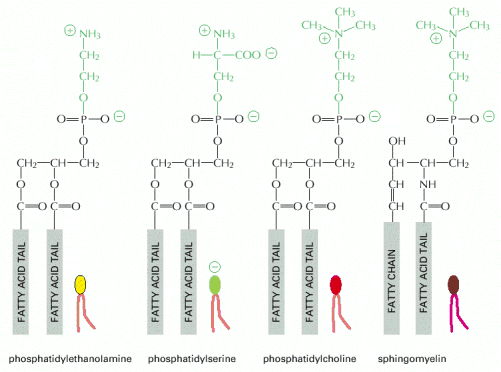{kind=link}
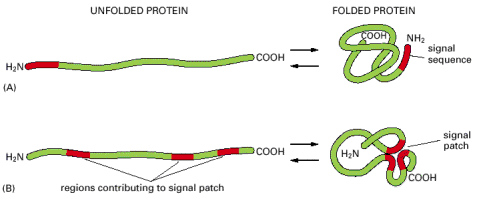{kind=link}
{kind=link}
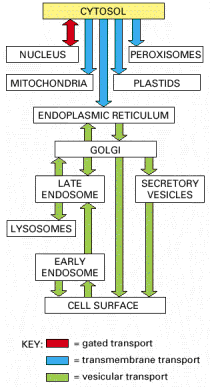{kind=link}
{kind=link}
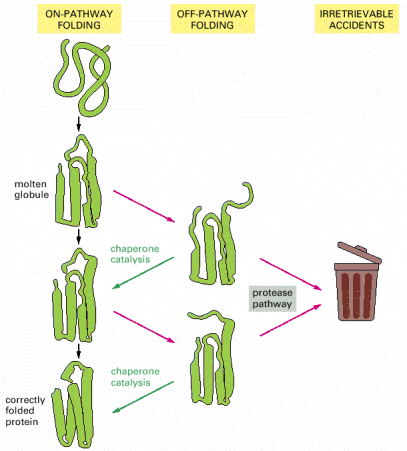{kind=link}
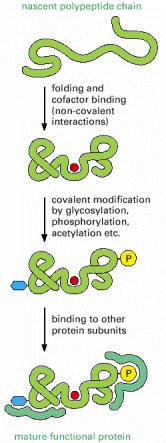{kind=link}
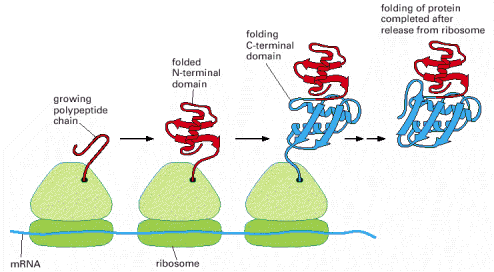{kind=link}
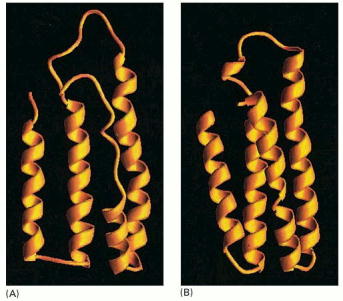{kind=link}
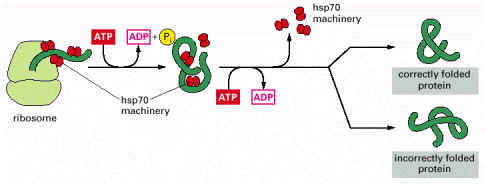{kind=link}
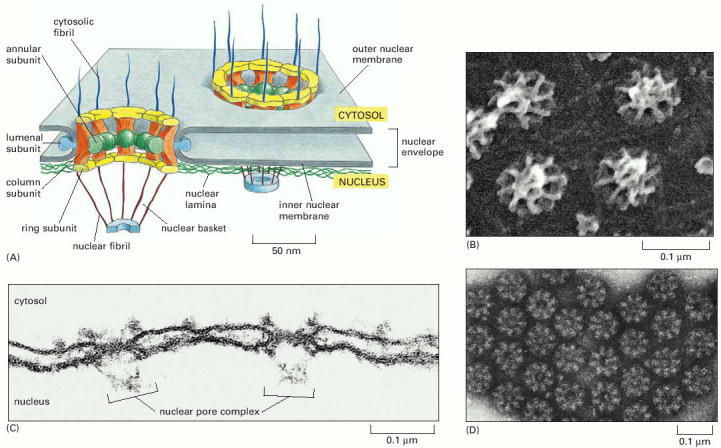{kind=link}
{kind=link}
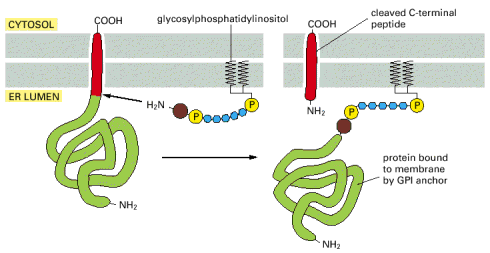{kind=link}
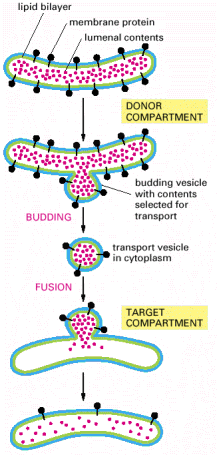{kind=link}
{kind=link}
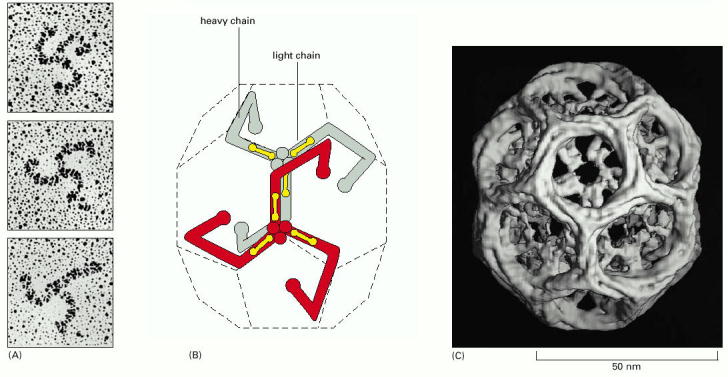{kind=link}
{kind=link}
{kind=link}
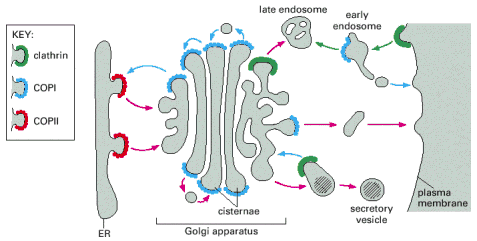{kind=link}
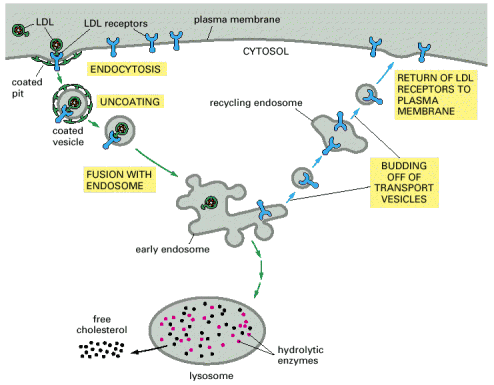{kind=link}
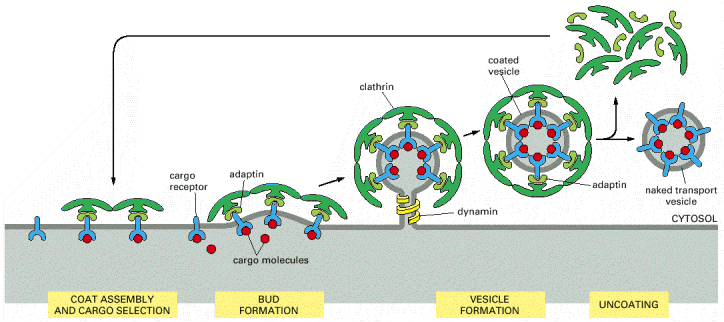{kind=link}
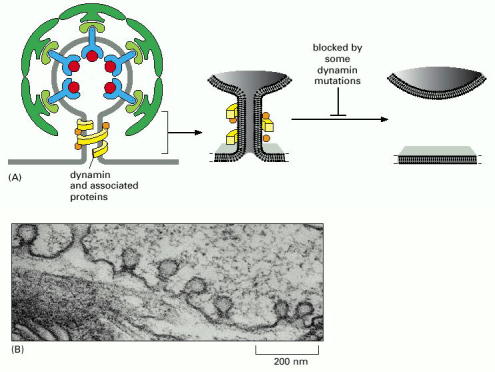{kind=link}
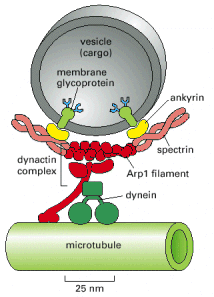{kind=link}
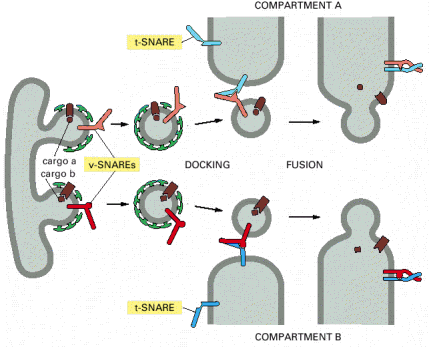{kind=link}
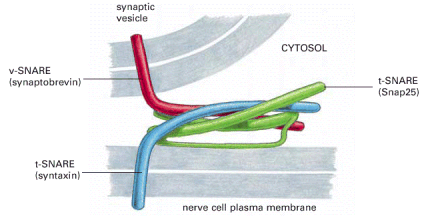{kind=link}
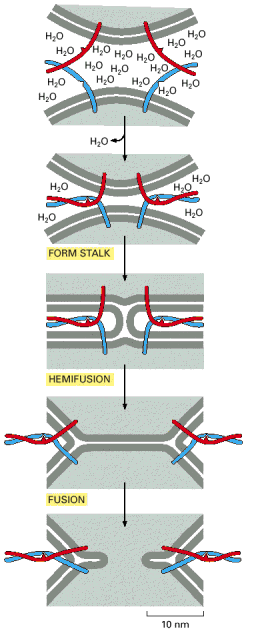{kind=link}
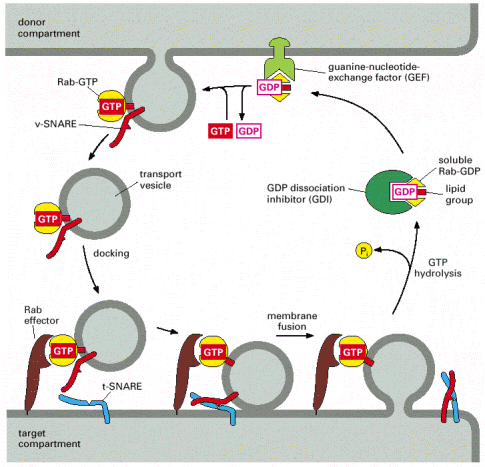{kind=link}
{kind=link}
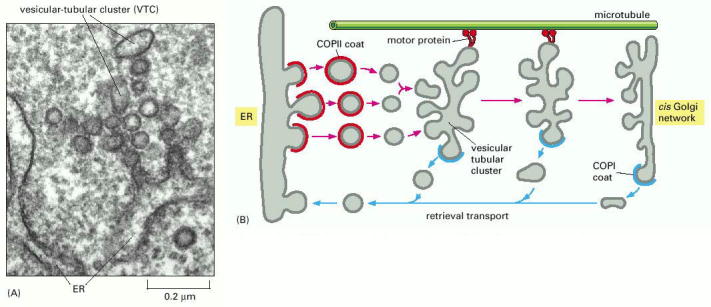{kind=link}
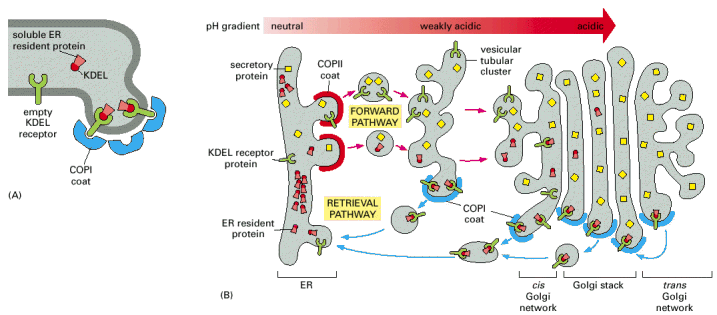{kind=link}
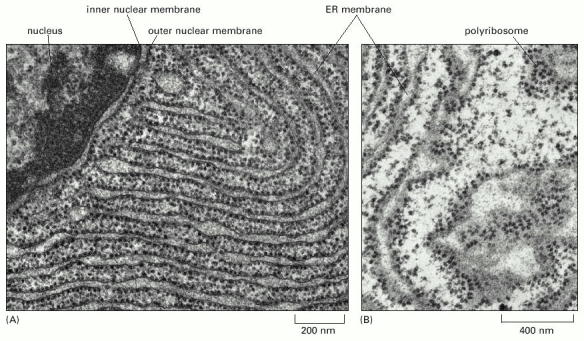{kind=link}
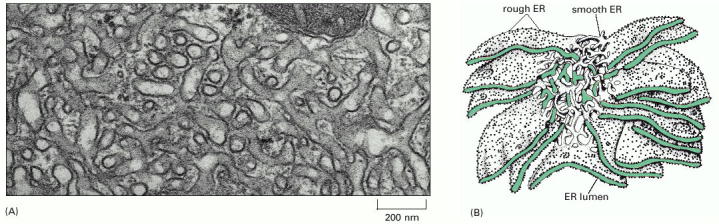{kind=link}
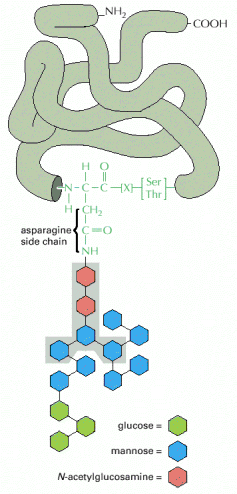{kind=link}
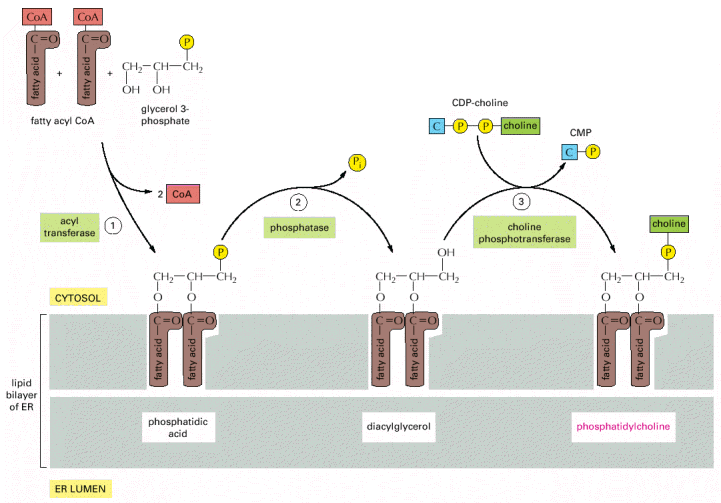{kind=link}
{kind=link}
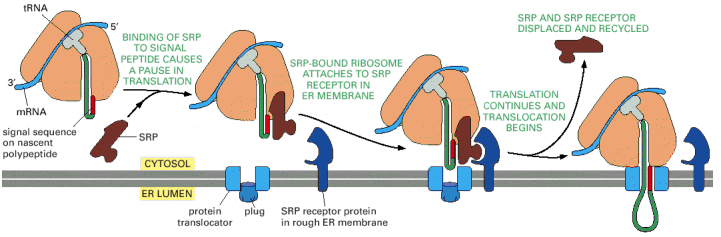{kind=link}
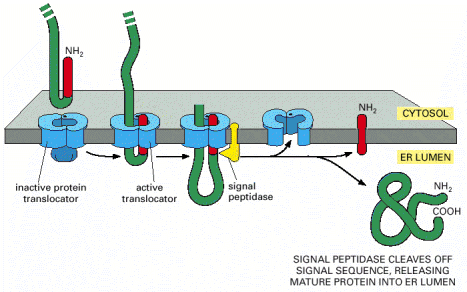{kind=link}
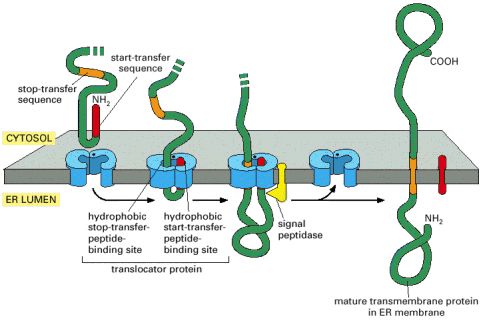{kind=link}
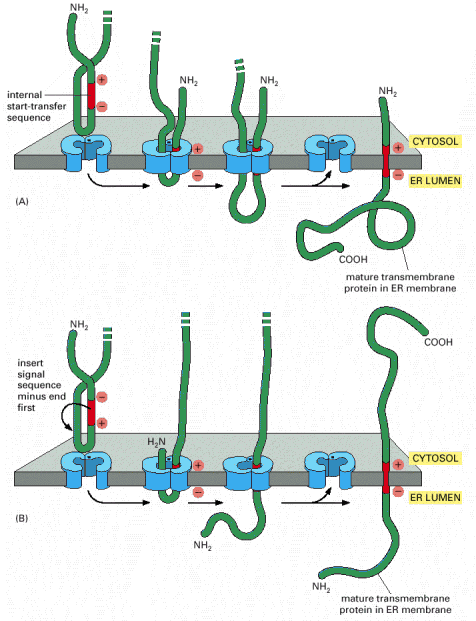{kind=link}
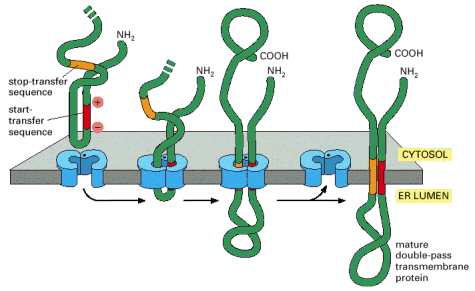{kind=link}
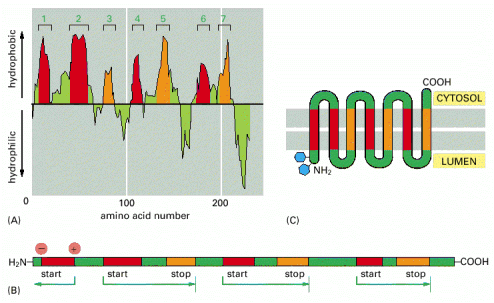{kind=link}
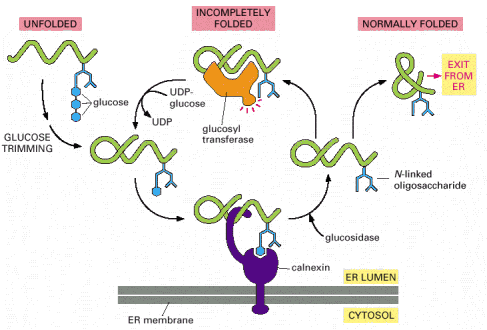{kind=link}
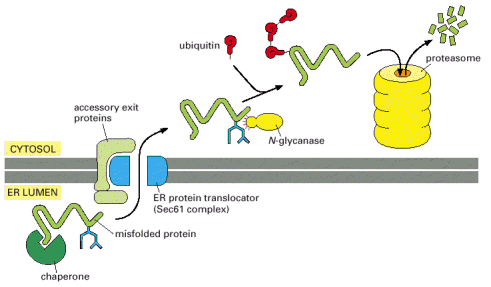{kind=link}
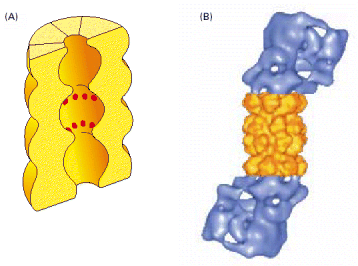{kind=link}
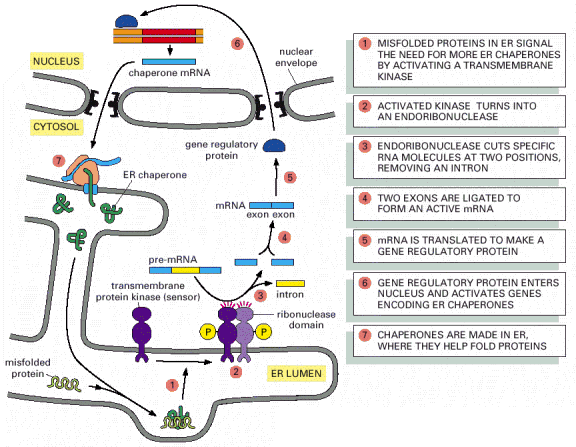{kind=link}
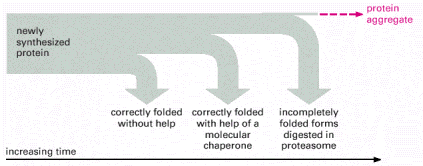{kind=link}
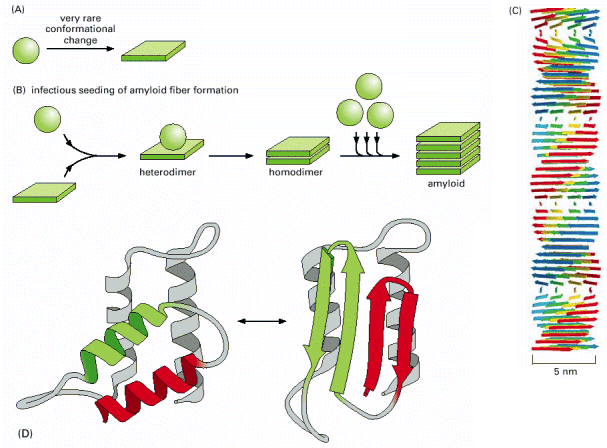{kind=link}
{kind=link}
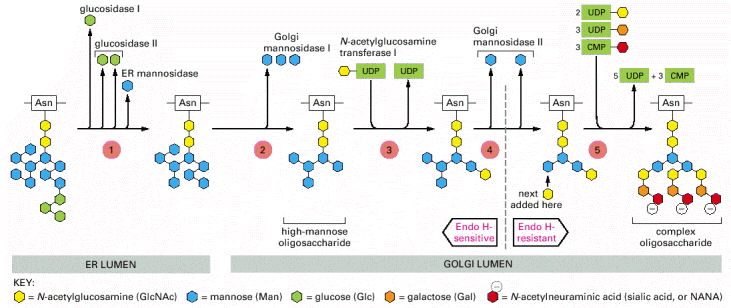{kind=link}
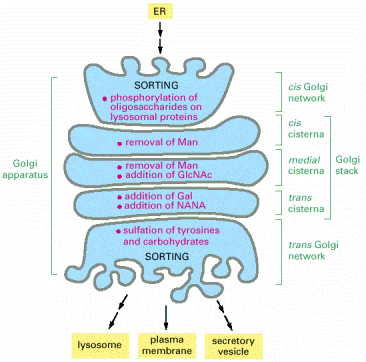{kind=link}
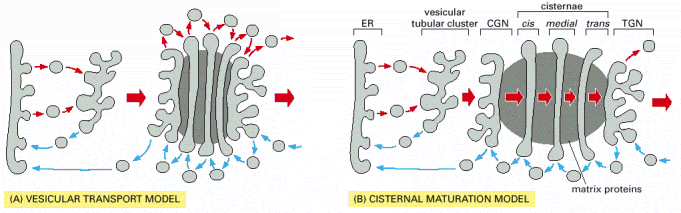{kind=link}
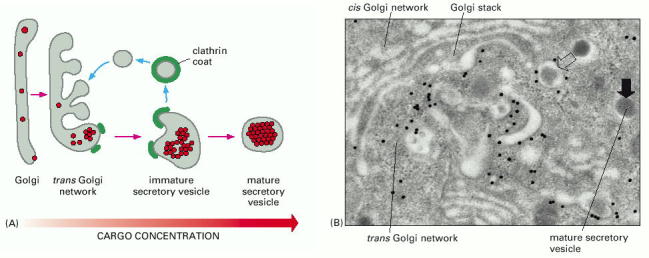{kind=link}
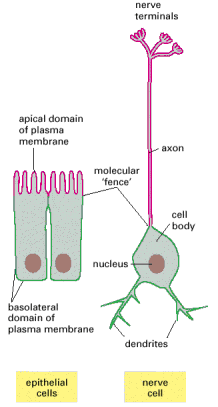{kind=link}
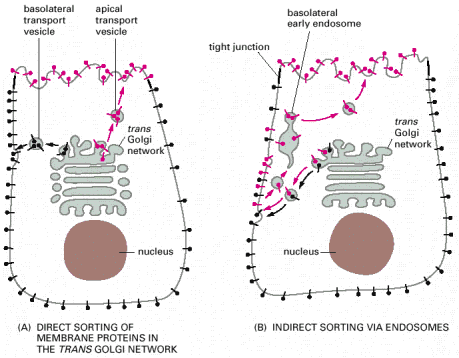{kind=link}
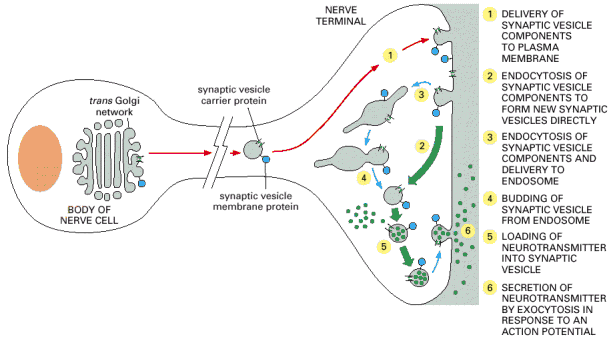{kind=link}
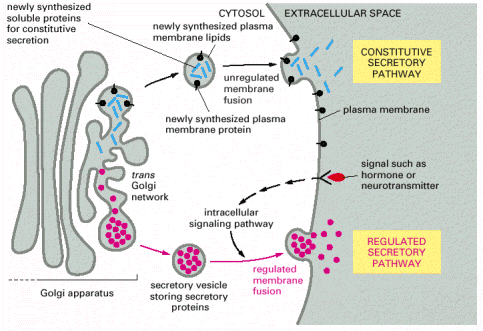{kind=link}
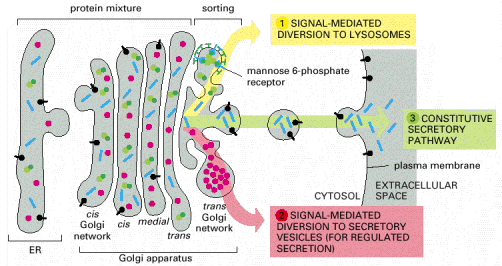{kind=link}
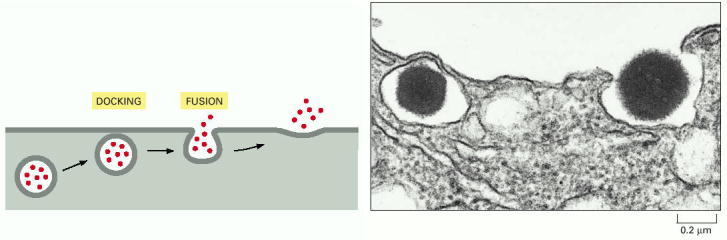{kind=link}
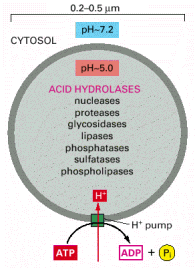{kind=link}
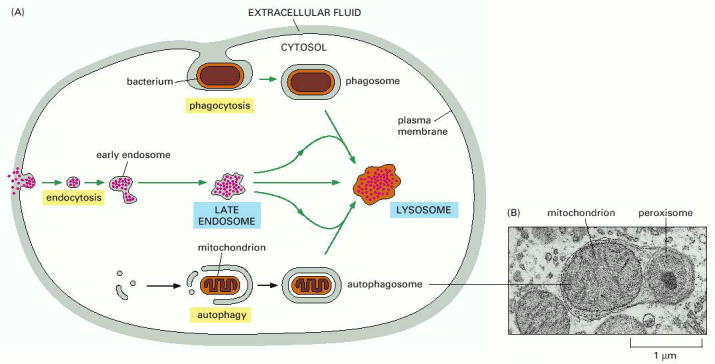{kind=link}
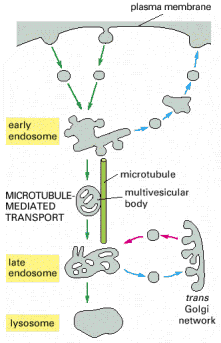{kind=link}
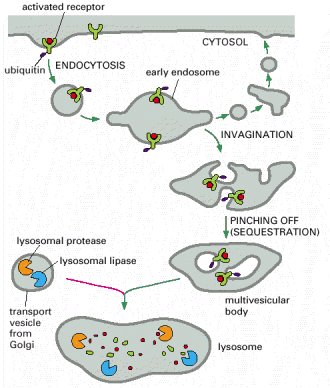{kind=link}
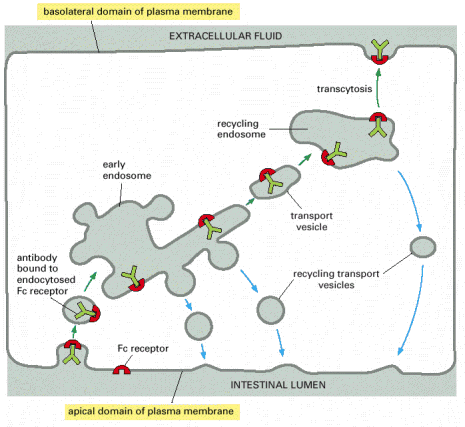{kind=link}
{kind=link}
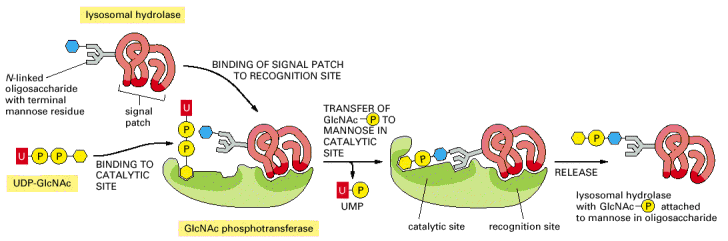{kind=link}
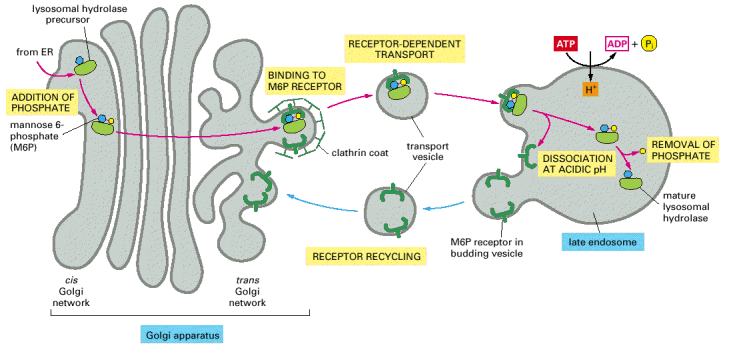{kind=link}
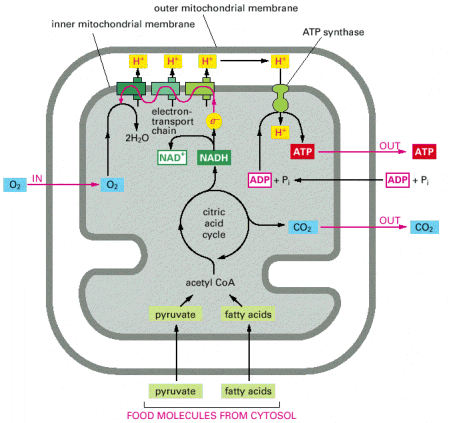{kind=link}
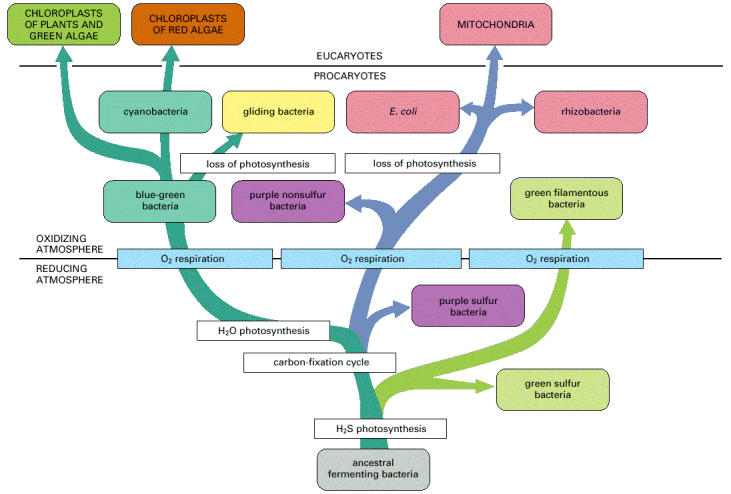{kind=link}
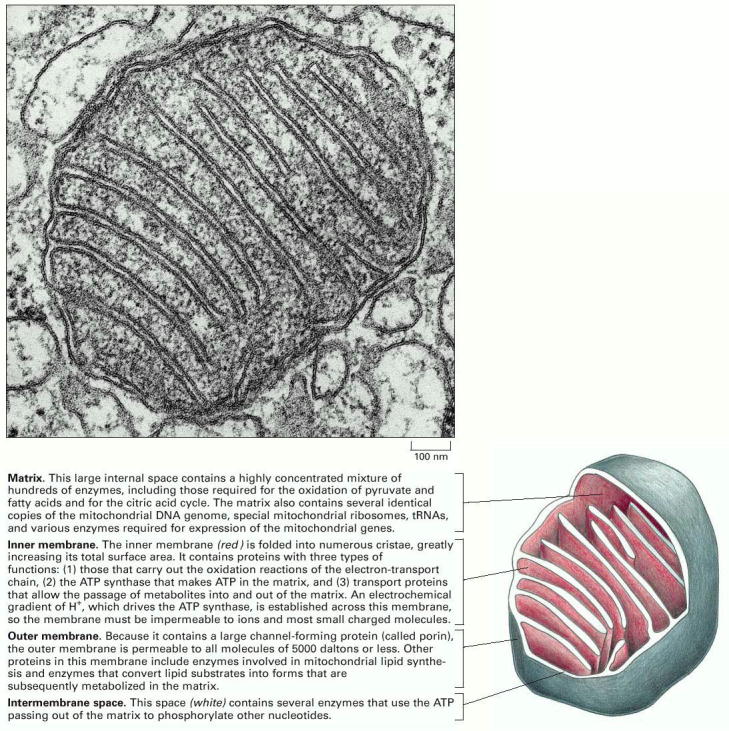{kind=link}
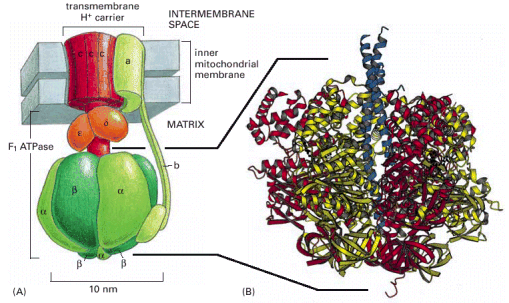{kind=link}
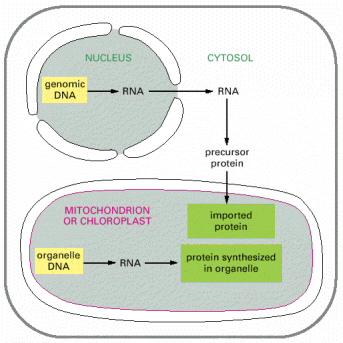{kind=link}
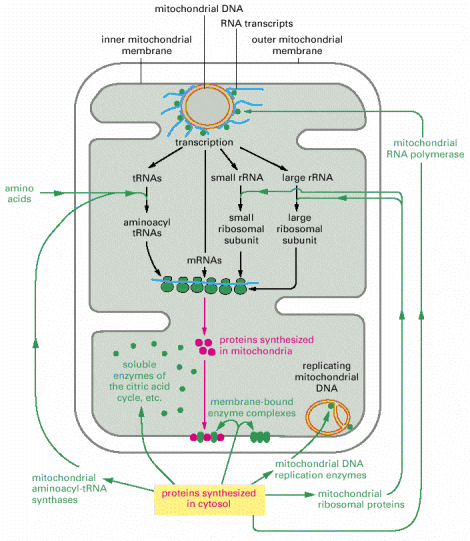{kind=link}
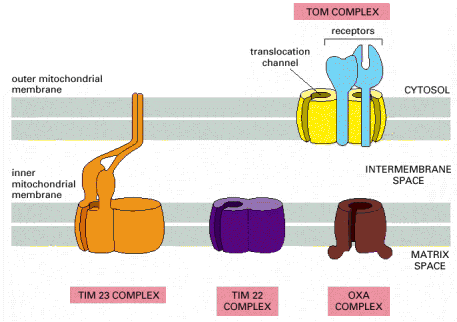{kind=link}
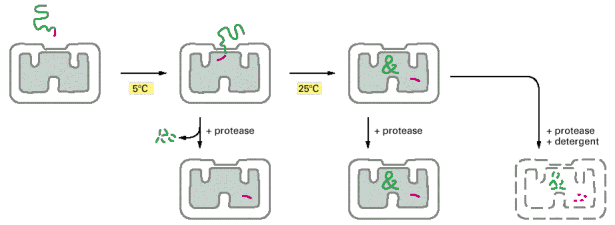{kind=link}
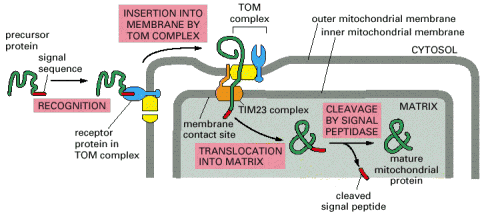{kind=link}
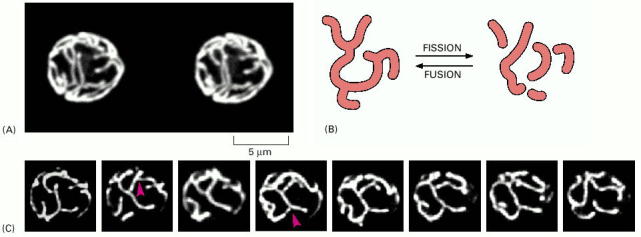{kind=link}
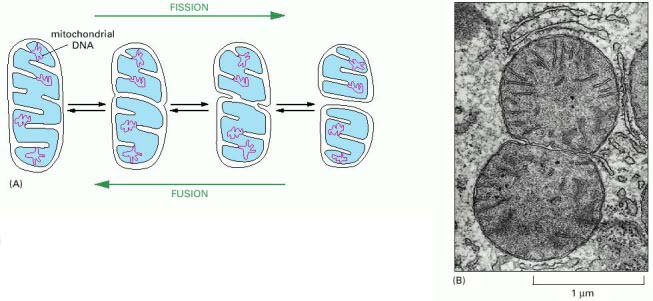{kind=link}
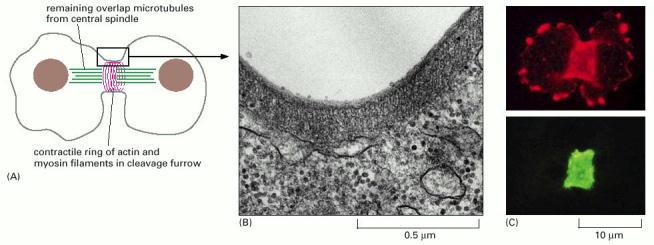{kind=link}
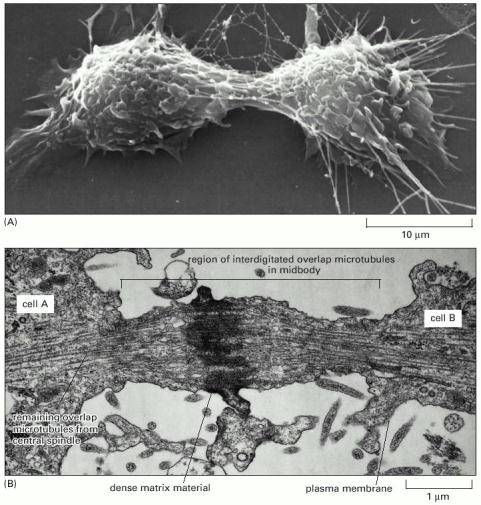{kind=link}