PDF verze textu
PDF verze obrázků
PDF verze schémat
Prof. MUDr. Milan Elleder, Dr.Sc., MUDr. Jakub Sikora, Ph.D. - Ústav dědičných metabolických poruch 1. LF UK a VFN
kapitola "Možnosti testování funkce lysosomálního systému in vivo v buněčných kulturách" - RNDr. Jana Ledvinová, CSc. - Ústav dědičných metabolických poruch 1. LF UK a VFN
kapitola "Siderosomy" - Přemysl Poňka, MD, PhD - Department of Physiology, McGill University, Montreal, Quebec, Canada
kapitola "Role ELS v imunitě" - MUDr. Zora Mělková, Ph.D. - Ústav imunologie a mikrobiologie 1. LF UK a VFN
Koncem 19. století byla publikována první klinická pozorování (Dr. Warren Tay, Dr. Bernard Sachs, Dr. Philippe C. E. Gaucher) závažných a progresivních vrozených onemocnění (později označených za m.Tay-Sachs a m.Gaucher) provázených těžkou neurologickou symptomatologií a zvětšením viscerálních orgánů (zejména sleziny), která byla v následném století zařazena mezi tzv. lysosomální střádací onemocnění.
P.C.E. Gaucher: De l´epithelioma primitiv de la rate. (These de Paris, 1882, (these studenta mediciny))
W. Tay: Symmetrical changes in the region of the yellow spot in each eye of an infant. (Trans.Ophthalmol.Soc. 1:155, 1881)
B. Sachs: A family form of idiocy, generaly fatal associated with early blindness. (J.Nerv.Ment.Dis. 21:475, 1896)
Klinická pozorování celé řady dalších, podobně závažných a v určitých aspektech, vzájemně si podobných patologických stavů, se objevila v průběhu první poloviny 20. století. V této době nemohla být tato onemocnění spojována s poruchou lysosomálního systému, který ještě nebyl objeven. Nicméně biochemici přispěli poznáním, že se v postižených orgánech, mnohdy výrazně zvětšených (organomegalie), akumulují (střádají) lipidy, takže vznikl obecný pojem "střádací onemocnění", což je dnes vysvětlitelné tím, že se dané látky hromadí ve vymezeném buněčném kompartmentu a nedifundují tělesnými tekutinami, jak je tomu u mnoha jiných geneticky podmíněných metabolických poruch.
Vlastní historie lysosomálního systémuV roce 1974 byla udělena Nobelova cena Dr. Christianovi DeDuveovi za dřívější objev, jak sám uvedl ve svém inauguračním projevu, "membrane endowed intracellular granules rich in hydrolytic enzymes". Tato granula obdaná membránou a bohatá na hydrolytické enzymatické aktivity pojmenoval lysosomy ("lytická tělíska"). Tato definice lysosomů vznikla v roce 1955 a vycházela ze strukturálních poznatků transmisní elektronové mikroskopie a biochemických měření aktivit hydrolytických enzymů po předchozí frakcionaci buněčných organel. Kombinace přístupů morfologických a biochemických dovolila definovat buněčnou organelu.
C. de Duve et al.: Tissue fractionation studies 6. Intracellular distribution patterns of enzymes in rat-liver tissue. (Biochem. J. 60: 604-617, 1955)
S. Goldfischer, E. Essner,A.B. Novikoff: The localization of acid phosphatase activities at the level of ultrastructure.( J. Histochem. Cytochem. 12: 72-95, 1964)
Po tomto "velkém třesku" se postupně rozvinuly znalosti o struktuře a funkci lysosomálního systému do neobyčejné šíře. Poznatky byly obohaceny i cíleným studiem genetických poruch lysosomálního systému, díky kterému byly identifikovány proteiny, jejichž funkce je doposud málo známa a které jsou předmětem intensivních studií. V tomto textu je podán přehled současného stavu poznání lysosomální biologie a patologie bez ambice o prezentaci všech známých molekulárních detailů a v současnosti ověřovaných hypotéz. Text by měl pomoci překlenout stále se rozšiřující mezeru mezi praktickou medicínou a základním výzkumem. Je k disposici všem, kteří mají zájem a motivaci udržet si kontakt s pokrokem v této oblasti biomediciny.
Začátek moderní klinické historie lysosomálních poruchK propojení obecně biologické znalosti o existence intracelulárního membránového kompartmentu bohatého na hydrolytické enzymové aktivity s realitou klinicky popsaných, ale kauzálně nepochopených klinických fenotypů střádání, došlo v roce 1963, kdy byl asociován fenotyp Pompeho nemoci s enzymatickým deficitem lysomální α-glukosidasy. Ten samý rok byla podobná asociace prokázána v případě tzv. metachromatické leukodystrofie (deficit enzymatické aktivity lysosomální arylsufatasy A).
H.G. Hers: Alpha-glucosidase deficiency in generalized glycogen storage disease (Pompe´s disease).(Biochem.J. 86: 11-16, 1963)
J.H. Austin et al.:A controlled study of enzymatic activities in three human disorders of glycolipid metabolism. (J.Neurochem. 10:805, 1963)
Ke dnešnímu dni bylo takto asociováno více než 50 dědičných onemocnění (a to až na úroveň DNA defektu), jejichž společným jmenovatelem je nějaká forma postižení lysosomální biologie (ne vždy v souvislosti s biochemickým deficitem enzymatické aktivity), provázená, ve větší či menší míře, rozvojem buď lysosomálního střádání, nebo procesu morfologicky podobného. V roce 2011 jsme schopni, na rozdíl od konce 19. století, a to je jistě zásadní pokrok, výše uvedený počet lysosomálních střádacích onemocnění definovat a precizně diagnostikovat (i prenatálně) na celé škále úrovní (klinických, zobrazovacích, histomorfologických, biochemických a molekulárně genetických).
Co lze ještě v oblasti patologie lidských lysosomálních střádacích onemocnění studovat, když je vše základní definováno? Komplexní patogenezi těchto stavů, což je nutný předpoklad k dalšímu rozvoji kauzální terapie! Stále tedy platí: "Vše v lidské patologii již bylo popsáno, zbývá už jen pochopit podstatu definovaného"
AFCS - acquired foam cell syndrome; AMPA - α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid; AMRF - action myoclonus renal failure syndrome; AP - adaptorový protein; APC - antigen presenting cells; ASC - apical sorting compartment; Atg - autophagy related genes; ATP7B - ATPase 7B; BLOC - biogenesis of lysosomes-related organel complexů; CESD - cholesteryl ester storage disease; CMA - chaperon mediated autophagy (chaperony zprostředkovaná autofagie); COPII -coatomer protein II; CURL - compartment of uncoupling of receptors and ligands; DMP - dědičné metabolické poruchy; DMT 1 - divalent metal transporter 1; ECV - endocytotic transport vesicles; ELS - endosomální lysosomální systém; EM - elektronová mikroskopie; ER - endoplasmatické retikulum; GB3 - globotriaosylceramid; GSD type II - glycogen storage disease type II; HE - barvení hematoxylinem eosinem; HPS - Heřmanský-Pudlák syndrom; HFE - high iron Fe; HO1 - hemoxygenasa 1; Ig - imunoglobuliny; LAMP2 - lysosomal associated membrane protein 2; LC3B-I a II - microtubule-associated protein light chain 3; LIMP2 - lysosomal integral membrane protein 2; LRO - lysosomal related organelles; LYST protein - lysosomal trafficking protein; M-6-P - manosa-6-fosfát; MALS - macroautophagy/lysosomal systém; MHC - main histocompatibility complex; ML - mukolipidosa; M-P - manosa-fosfát; MPR - M-6-P receptor; MPR46/CD-MPR - kation dependentní M-6-P receptor o molekulové hmotnosti 46 kDa; MPR300/CI-MPR - kation independetní M-6-P receptor o molekulové hmotnosti 300 kDa; MPS - mucopolysaccharidosis (mukopolysacharidosa); NB-DNJ - N-butyl-deoxyjonojirimycinem; NCL - neuronální ceroid lipofuscinosa; NPC - Niemann-Pickova choroba typ C; NRAMP 1 - natural resistance macrophage-associated protein 1; OXFOS - oxidativní fosforylace; PAF - permanganat-aldehydfuchsin; PAS - perdiodic acid Schiff; PPT - palmitoyl protein thioesterasa; PSAP - prosaposin; RARS - refractory anemia with ring sideroblasts; Rab -Rab - GTPase (rabbit-like); RER - rough endoplasmic reticulum; RNAse t2 (RNASET2) - ribonukleasa T2; SAP - saposin - sphingolipid activator proteins; SNARE -soluble n-ethyl maleimide sensitive attachment proteins receptor; SRT - substrate reduction therapy; TAP - transporter associated with antigen processing; TFEB - transcription factor EB; TPP1 - tripeptidyl peptidasa 1; UPS - ubiquitin proteasomový systém
Definici lysosomů Christiana De Duvea z roku 1955 je třeba s ohledem na stav současného poznání modifikovat. Lysosomy jsou zajisté stále "membránou obdaná granula s vysoký obsahem hydrolytických enzymů". To, co se změnilo, je objem informací svědčících pro skutečný rozsah dynamiky lysosomálního membránového systému a vztahů lysosomů k jiným intracelulárním kompartmentům či signalizačním drahám. V souvislosti se skutečností, že hlavní, nikoliv však jedinou, cestou substrátového toku do lysosomů je endocytosa, jsou lysosomy logicky funkčně spojovány zejména s touto drahou, proto označení - endosomálně lysosomální systém (ELS). V rámci kaskády endocytosy jsou lysosomy finální degradativní etáží, která velice úzce komunikuje s úrovní pozdního endosomu, jenž je skutečnou integrativní úrovní ELS (viz níže), kde se stýká nově syntetizovaný funkční proteom směřovaný z Golgiho aparátu se všemi vstupními substrátovými drahami (endocytosa, fagocytosa, autofagocytosa).
Koncept ELS představuje dynamický, mnohočetný a několikaetážový cytoplasmatický vakuolární kompartment přítomný ve všech buňkách. Historicky nejdéle známou funkcí je funkce degradační, a to jak látek přicházejících do buňky z extracelulárního prostoru (endocytosa a fagocytosa), tak i substrátů intracelulárních (autofagocytosa). Tímto způsobem získávají buňky zdroje nejen pro vlastní metabolismus, ale zároveň se významně podílejí na regulaci celé řady extracelulárních komponent (např. sérových proteinů nebo extracelulární matrix). ELS se dále významně podílí na degradační části obratu cytoplasmatických komponent včetně buněčné membrány. Spolu s proteasomem (UPS) se účastní intracelulární degradace chybných proteinových konformerů. Všechny tyto degradační hydrolytické procesy jsou realisovány baterií několika desítek kyselých hydrolas (viz níže), jejichž funkcí je rozklad biokonjugátů, přičemž produkty těchto reakcí jsou recyklovány.
Koordinace toku luminálního a membránového materiálu v rámci ELS je nesmírná a v řadě aspektů stále nedostatečně prostudovaná. ELS významným způsobem spoluzajišťuje prostřednictvím redistribuce a recyklace biologického materiálu základní buněčnou homeostasu. Velká část interakcí v rámci ELS může zahrnovat kontakty složitějších fázových rozhraní (hydrofilní-hydrofobní nebo hydrofobní-hydrofobní) anebo interakce protein-proteinové a protein-membránové. Právě s ohledem na míru a rozsah membránových a proteinových interakcí ELS v buňce, lze tento systém považovat za určitý centrální koordinátor zejména lipidního metabolismu ve smyslu degradace a recyklace resp. redistribuce. S vědomím faktu, že významnými lysosomální substráty z této skupiny jsou látky typu cholesterolu či gangliosidů, které jsou naprosto esenciální pro organizaci celé řady na membrány vázaných signalizačních drah, dostává intracelulární funkce ELS daleko širší rozměr jdoucí nad rámec původní definice Christiana DeDuvea. Zvláštní funkcí, kterou i tento text uvádí samostatně, je biogenese systému sekrečních organel blízkých lysosomům (LRO), který s ELS sdílí řadu molekulárních charakteristik.
Použití pojmů:Snahy o statickou popisnou stratifikaci ELS, který je ze své biologické podstaty enormně proměnlivý svou mnohosměrností transportu nákladů a vlastních biologických membrán, mohou vést k nechtěným a problematickým zjednodušením a obsahovým zkratkám. Samotné označení ELS je z podstaty nepřesné, jelikož odráží pouze jeden (sice kvantitativně nejvýznamnější) ze základních toků substrátů určených k degradaci. Obecný koncept rozdělující ELS na definované vesikulární populace (časné, recyklující a pozdní endosomy a lysosomy, viz níže) by měl vždy být přijímán se znalostí reálné dynamiky a proměnlivosti celého tohoto membránového systému. Těsné funkční a prostorové propojení pozdně endosomálního kompartmentu (manosa-6-fosfát (M-6-P) positivní) a lysosomů ( M-6-P negativní) vedlo k zavedení často používaného pojmu - pozdní endosom(y)/lysosom(y) (late endosomes/lysosomes).
Tento text nemá za cíl věnovat se extenzivně časným fázím endocytosy, proto jsou pojmy ELS, pozdně endosomální/lysomální a lysosomální, používány v ekvivalentním smyslu.
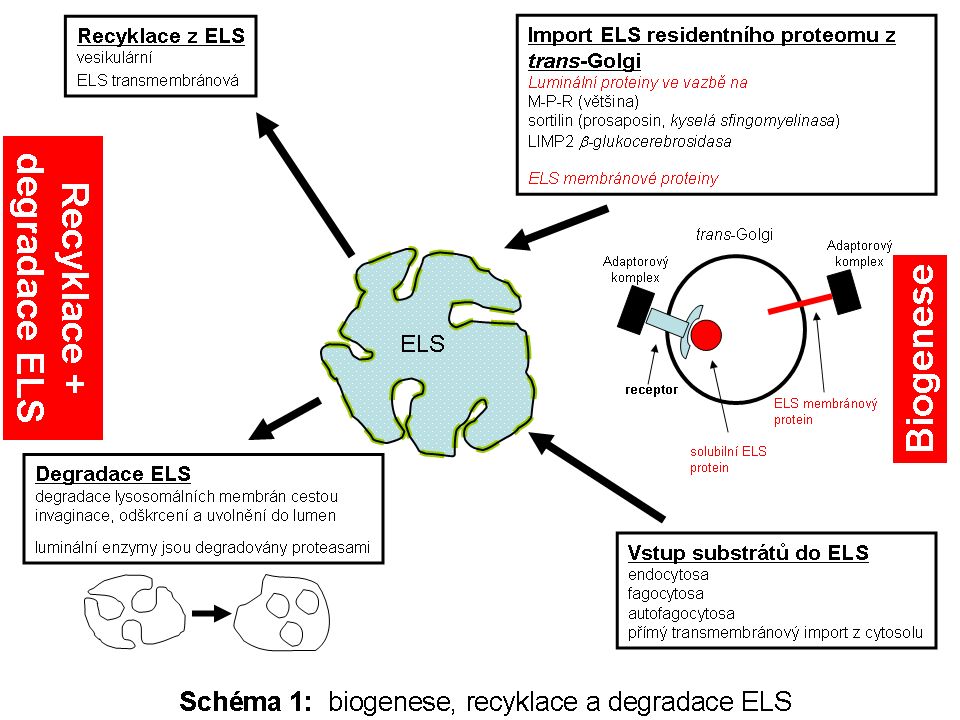
Komplexita biologického systému ELS bude v rámci tohoto textu vysvětlena na podkladě jeho biogenese, přičemž z podstaty funkce ELS není možné oddělit strukturální sebeobnovu této organely od biologie intracelulárních mechanismů a drah zajišťujících import substrátů určených k degradaci a recyklaci produktů této degradace. ELS je podobně jako ostatní membránové organely maternálního původu.
Proto, aby ELS fungoval jako jedno z degradačních a recyklačních center eukaryotické buňky, je třeba, aby byl ELS jako organela strukturálně ustaven, tzn. je nutná trvalá strukturně biologická obměna ELS proteomu cíleným vesikulárním transportem membránových i luminálních komponent z oblasti trans-Golgi po předchozí syntese v ER. Část ELS proteinů se může do ELS dostávat (jako forma specifické retence a zpětného zacílení) z jiných intracelulárních kompartmentů (např. z cytoplasmatické membrány) či extracelulárního prostoru (viz níže).
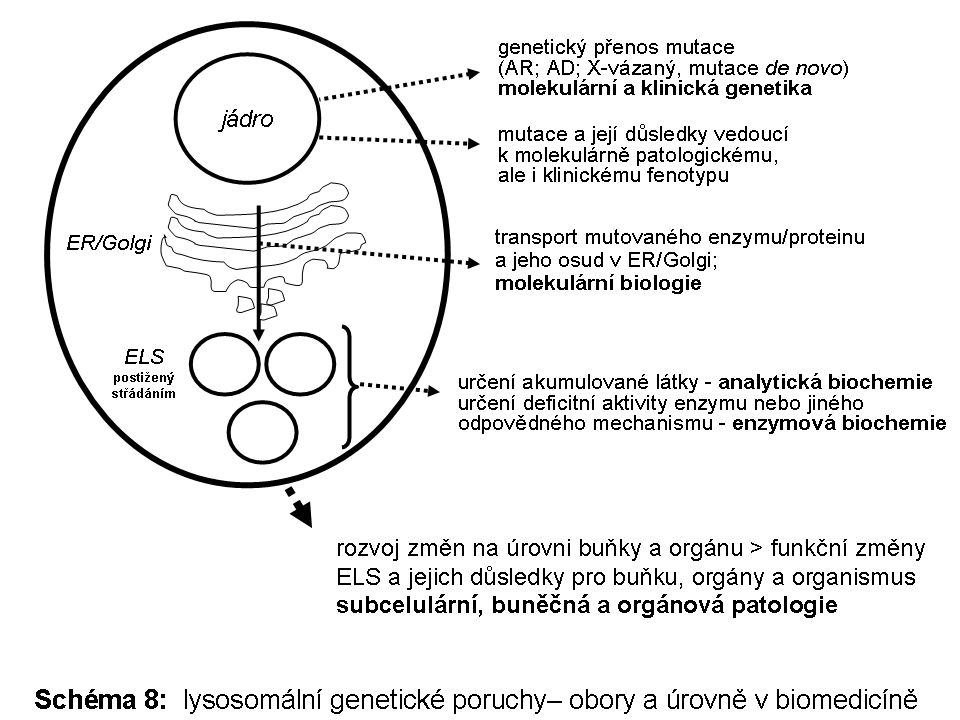
Biogenesí je myšlen souhrn všech mechanismů zajišťujících koordinovanou strukturální a proteomickou sebeobnovu ELS a zároveň tak souhrn drah zajišťujích přísun biologických substrátů určených k degradaci do ELS. Úzce jaou na tento systém dále napojeny dráhy určené k intracelulární recyklaci produktů subtrátové degradace z ELS (Schéma 1).
Proteom ELS (v detailu níže) lze pro zjednodušení obecně rozdělit na skupinu luminálních solubilních proteinů (drtivá většina lysosomálních hydrolas) a skupinu proteinů fungujících v lysosomální limitující membráně. Celá řada lysosomálních proteinů, byť solubilních, funguje v těsné asociaci s hydrofobními lipidickými fázemi membránové povahy, proto pro svou funkci velmi často potřebují zprostředkující aktivátor (v detailu níže).
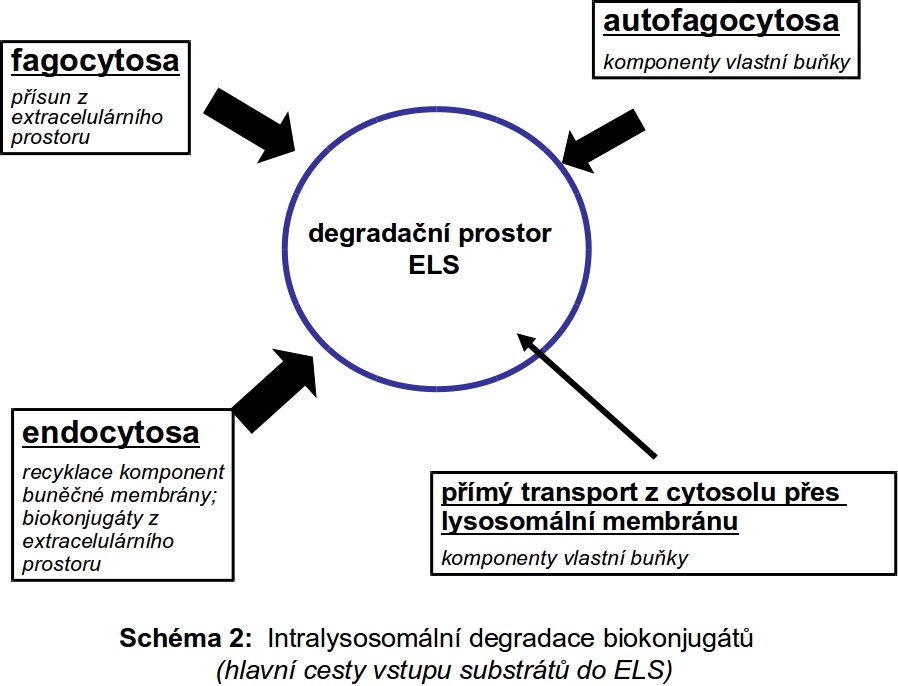
Biokonjugáty (substráty) určené k degradaci v ELS (Schéma 2) pocházejí z:
1. extracelulárního prostoru, ze kterého se dostávají do buňky mechanismem více či méně selektivní endocytosy a fagocytosy. Touto cestou je zpracován přísun vysokomolekulárních látek z extracelulárního prostoru, včetně buněk určených k likvidaci. Průnik látek do buněk je buď bez jakéhokoliv omezení, jako je tomu v případě nepolárních neutrálních nízkomolekulárních látek, nebo je striktně kontrolován proteinovými transportéry, lokalisovanými na buněčné membráně a specializovanými na přenos polárních molekul. Mezi těmito dvěma systémy transportu individuálních molekul přes membránu stojí proces vesikulárního transportu - endocytosy, který umožňuje průnik nejrůznějších biopolymerů do buňky v membránou ohraničeném prostoru endocytických váčků. Osud těchto váčků včetně jejich obsahu je různý. Kvantitativně nejvýznamnější a relativně dobře definovaná je jejich postupná degradace v ELS. Vedle funkce degradativní je zejména na časné fáze endocytosy vázána celá řada signalizačních intracelulárních drah, kterým se však v rámci tohoto textu nebudeme věnovat.
2. zdrojem biopolymerů určených k degradaci je vlastní cytoplasma, tzn. intracelulární prostředí. Fysiologickým degradativním mechanismem neustálé sebeobnovy cytoplasmy je autofagocytosa.
3.1.1. Strukturální a proteomická biogenese ELS (pozdních endosomů/lysosomů)
Historický pohled na lysosomální biologii dlouhou dobu akcentoval biochemické aspekty související s aktivitami solubilních lysosomálních hydrolytických enzymů. Současný stav poznání a náhledu na proteomiku ELS (myšleno pozdních endosomů/lysosomů) je nutno rozšířit především o skupinu integrálních membránových proteinů limitující lysosomální membrány. Většina proteinů z této skupiny nemá katalytickou funkci (i přesto jejich geneticky podmíněné defekty vedou k rozvoji některých lysosomálních střádacích onemocnění, viz níže). V této souvislosti považujeme za nutné uvést obecnější aspekty buněčné biologie syntesy, posttranslačních modifikací, a dále specifikovat způsoby intracelulárního cílení proteinů do ELS (pozdních endosomů/lysosomů).
Pozdně endosomální/lysosomální proteom savčích buněk zahrnuje přibližně 60-100 luminálních (solubilních nebo solubilních, ale s membránami asociovaných) hydrolas o různé substrátové specificitě (proteasy, nukleasy, glykosidasy, lipasy, fosfolipasy, fosfatasy a sulfatasy). Kyselé intraluminální pH pozdních endosomů/lysosomů nutné pro optimální enzymatickou funkci většiny těchto hydrolas je zajišťováno biologickou aktivitou tzv. vakuolární H+ATPasy a chloridovým proteinovým kanálem (v obou případech se jedná o rezidentní proteiny lysosomální limitující membrány).
Vedle luminálních hydrolas mají pozdní endosomy/lysosomy ještě membránový proteom čítající dalších 130-150 proteinů včetně již dříve uvedené H+ATPasy a Cl- kanálu. Mezi membránovými proteiny jsou nejhojněji zastoupené vysoce glykosylované lysosomálně asociované membránové proteiny (LAMP1 a 2) a lysosomální integrální membránové proteiny (LIMP). Vysoký stupeň glykosylace těchto proteinů s významným podílem obtížně degradovatelných polylaktosaminů může sloužit, mimo jiné, jako protektivní bariera chránící lysosomální limitující membránu před agresivními luminálními hydrolasami.
Pro úvodní shrnutí - celkový pozdně endosomální/lysosomální proteom (odhadované počty uvedeny v závorkách) je tvořen jak luminálními hydrolasami (60-100), tak lysosomálními membránovými proteiny (130-150).
Cílení proteinů do ELS (pozdních endosomů/lysosomů)Lysosomální hydrolasy a lysosomální membránové proteiny jsou syntetizovány na ribosomech vázaných na cisterny RER obecně známým způsobem (uvádíme pouze stručné shrnutí).
ER importní signální sekvence (kotranslačně kotvené do membrány RER) solubilních luminálních hydrolas jsou v nově syntetisovaných proteinech lokalisovány N-terminálně. Po skončení nebo někdy již v průběhu translace nového proteinu do cisteren RER jsou signální sekvence signální peptidasou odštěpeny (vyjímkou může být např. kyselá fosfatasa viz níže). U lysosomálních membránových proteinů mohou být tyto sekvence interní v závislosti na transmembránové orientaci individuálního proteinu. Lysosomální proteiny se v ER iniciálně sbalují (jsou konformovány do stabilních forem) s pomocí ER rezidentních proteinových chaperonových interakčních sítí. Sbalovány jsou nejen solubilní luminální proteiny, ale i extramembránové domény (obrácené do ER i do cytosolu) integrálních membránových proteinů. V ER jsou proteiny také iniciálně glykosylovány, což také souvisí s jejich konformačním vyzráváním. V případě dosažení stabilního konformačního stavu a po kontrole jeho kvality (znovu zajišťováno ER rezidentními proteinovými chaperony) opouštějí lysosomální proteiny ER skrze COPII výstupní místa v opláštěných váčcích (ER/Golgi intermediáty) směrem k cis-Golgi cisterně. Tento obecný mechanismus (ER residentní sbalování lysosomálních proteinů) se v dnešní době stal cílem jak experimentálních terapeutických přístupů tak i klinických terapeutických aplikací (viz níže).
Na tomto místě stojí za zmínku specifická posttranslační úprava celé skupiny lysosomálních sulfatas (arylsulfatasa A a B, iduronát 2-sulfatasa, sulfamidasa, galaktoso-6-sulfatasa, N-acetylgalaktosamin-4-sulfatasa a glukosamin sulfatasa) v ER vytvořením jejich společného aktivního centra (enzymová přeměna cystinu na formylglycin). Molekulárně genetický dědičný defekt proteinu odpovědného za tento typ posttranslační modifikace je podkladem tzv. polysulfatasového deficitu (multiple sulphatase deficiency, viz níže).
V cisternách Golgiho aparátu jsou pozdně endosomální/lysosomální proteiny glykosylovány, a po výstupu z trans- Golgi cisterny jsou vesikulárním transportem cíleny do pozdních endosomů, kde se setkávají, v případě luminálních hydrolas, se svými substráty (viz níže). Transport probíhá ve vazbě na transmembránový receptor (viz níže). Některé lysosomální proteiny mohou vytvářet ještě aditivní transportní komplexy, na příklad se může jednat o komplex β-galaktosidasy, neuraminidasy, N-acetylgalaktosamin 6-sulfát sulfatasy a protektivního proteinu/katepsinu A.
Jak již bylo zmíněno, většina solubilních luminálních lysosomálních hydrolas je v Golgiho aparátu dále posttranslačně glykosylována či sulfatována. Další významnou, v tomto případě post-Golgi, posttranslační modifikací je intralysosomální proteolytické štěpení celé řady lysosomálních hydrolas, typicky se jedná třeba o štěpení prekurzorové molekuly prosaposinu (viz níže).
Ve smyslu glykosylace jsou solubilní ELS hydrolasy modifikovány N-vázanými oligosacharidovými řetězci s vysokým obsahem manosy. Nejvýznamnějším krokem celé sekvence glykosylačních posttranslačních modifikací solubilních lysosomálních proteinů je syntesa manosa-6-fosfátových (M-6-P) značek, která probíhá ve dvou po sobě jdoucích enzymatických krocích. V prvním kroku jsou lysosomální enzymy fosforylovány N-acetylglukosamin-1-fosfátem na C6-hydroxylových skupinách některých manosových zbytků. Katalysatorem této reakce je enzym UDP(uridindifosfát)-N-acetylglukosamin-1-fosfotransferasa (fosfotransferasa). V druhém kroku je N-acetylglukosamin odštěpen enzymem N-acetylglukosamin-1-fosfodiester α-N-acetylglukosaminidasou (fosfoglykosidasa). Tato druhá reakce odhalí M-6-P značku, která tak může být rozpoznána MPR v mebránách trans-Golgi aparátu. K M-6-fosforylaci dochází na ELS proteinech predilekčně v oblastech s koncentrací 2-3 lysinových zbytků ve vzájemné vzdálenosti 34 Å. Multiplikace M-6-P značek na jedné molekule lysosomálního proteinu zvyšuje afinitu pro M-6-P receptory - MPR (taktéž využíváno v terapeutických aplikacích, viz níže). Geneticky podmíněný deficit této části glykosylační sekvence (tvorba M-6-P značky) lysosomálních proteinů je příčinou mukolipidosy typ II (viz níže).
MPR váží lysosomální hydrolasy svými luminálními doménami orientovanými do trans- Golgi cisterny. Transportní váčky nesou tyto proteinové komplexy do pozdních endosomů, kde vlivem nižšího pH komplex enzym/MPR disociují, a receptory jsou recyklovány zpět do trans-Golgi nebo na cytoplasmatickou membránu (za účasti nedávno identifikovaného proteinu retromeru). Přítomnost MPR na cytoplasmatické membráně zajišťuje vychytávání lysosomálních proteinů, které se dostaly do extracelulárního prostoru. Jeden ze způsobů, jak se ELS proteiny nesoucí M-6-P značku do extracelulárního prostoru mohou dostat, je regulovaná exocytosa lysosomů (viz níže). Druhým možným mechanismem je konstitutivní exocytosa M-6-P proteinů z trans-Golgi.Předpokládá se, že ~ 5-20% lysosomálních proteinů unikne specifické vazbě na MPR v trans-Golgi.
Biologie MPR je velmi komplexní. Byly identifikovány dva typy MPR: kation dependentní MPR o molekulové hmotnosti 46 kDa (MPR46/CD-MPR) kation independetní MPR o molekulové hmotnosti 300 kDa (MPR300/CI-MPR). MPR46/CD-MPR má jedno M-6-P vazebné místo zatímco MPR300/CI-MPR má 2 M-6-P vazebná místa. MPR jsou v buňce lokalisovány zejména v trans-Golgi aparátu, endosomech a na cytoplasmatické membráně (zde konkrétně 3-10% celkového buněčného MPR). Naopak MPR nejsou přítomny díky recyklaci do trans-Golgi v lysosomech (viz výše).
Pouze MPR300/CI-MPR funguje na cytoplasmatické membráně jako receptor pro internalizaci extracelulárních M-6-P proteinů (využíváno v terapii lysosomálních střádacích onemocnění, viz níže). MPR300/CI-MPR má ještě další ligandy (např. insulin like growth factor II, retinová kyselina a další).
Transportu mezi trans-Golgi a ELS se účastní oba typy MPR a to pro všechny M-6-P značené ELS proteiny. Existují však určité molekulárně preferované interakce - MPR46/CD-MPR + RNAse t2 nebo heparanasa a MPR300/CI-MPR + α-manosidasa nebo katepsin D.
Velmi významným, a znovu, doposud nedořešeným bodem ELS biogenese je znalost mechanismů odštěpování M-6-P značky takto dříve modifikovaných proteinů. Skutečností je, že M-6-P deglykosylace neprobíhá ve všech buněčných typech stejně intensivně, dokonce lze předpokládat, že ELS systém individuální buňky může být v tomto smyslu funkčně stratifikován (viz níže).
Některé lysosomální hydrolasy jako katepsin B, kyselá alfa-glukosidasa nebo lysosomální kyselá fosfatasa jsou transportovány do pozdních endosomů/lysosomů ve svých prekurzorových formách a dochází na nich k autokatalytickým či jinak zprostředkovaným (např. jinými residentními proteasami) proteolytickým štěpením do aktivních forem.
Zajímavým a v mnoha ohledech netypickým je případ lysosomální kyselé fosfatasy, která je jedním z nejdéle známých lysosomálních proteinů ze skupiny hydrolas. Lysosomální kyselá fosfatasa je kódována genem ACP2 (lokalisován na chromosomu 11) a je iniciálně syntetisována jako transmembránový protein. Do ELS je cílena nepřímo přes cytoplasmatickou membránu. V průběhu cílení tohoto proteinu do ELS dochází k sérii proteolytických posttranslačních úprav. Prvý odštěpený peptid u kyselé fosfatasy je v části enzymové molekuly, orientované do cytosolu. Druhou úpravou je odštěpena část orientovaná do lumen lysosomu. Touto druhou úpravou se stává z pevně transmembránově vázaného enzymu volný solubilní enzym. Paradoxně není funkce kyselé fosfatasy plně objasněna. Vedle uvedené lysosomální kyselé fosfatasy existuje ještě (jako jeden z větší skupiny) tzv. tartarátrezistentní kyselá fosfatasa neboli uteroferrin. Jedná se o ELS protein, který je kódován genem ACP5 (lokalisován na chromosomu 19), a v ELS, podle všech dostupných informací, katalysuje (nemusí být jediný) odštěpování M-6-P značek. Přítomnost tohoto proteinu v rámci ELS není homogenní, což alespoň částečně vysvětluje intracelulární variabilitu v míře M-6-P deglykosylace ELS residentních proteinů.
Vedle MPR cesty cílení lysosomálních proteinů existují ještě alternativní formy selektivního transportu proteinů do tohoto buněčného kompartmentu. Prosaposin, prekurzor čtyř lysosomálních saposinů (A-D), kyselá sfingomyelinasa, GM2 aktivátorový protein a katepsiny A a H sdílejí společný intracelulární receptor sortilin pro cílení do pozdních endosomů.
β-glukocerebrosidasa využívá pro své cílení do pozdních endosomů/lysosomů lysosomální integrální membránový protein 2 (LIMP2). Genetický deficit LIMP2 má tedy, mimo jiné, také parametry lysosomální absence β-glukocerebrosidasy (viz níže).
Aktivátory lysosomálních enzymů. Pro správnou funkci lysosomálních enzymů štěpících sfingolipidy jsou nezbytné tzv. saposiny (odvozeno od SAP - sphingolipid activator protein), polypeptidy nutné k dokonalé interakci substrátu s enzymem. Jsou známy čtyři hlavní saposiny (A-D), syntetisované ve formě společného prekursoru prosaposinu (PSAP). Jsou více či méně specifické pro některé ze sfingolipidhydrolas (např. saposin C pro β-glukocerebrosidasu, saposin D pro ceramidasu). Genetické deficity PSAP a jednotlivých SAP proteinů jsou popsány níže. Specifické cílení PSAP do pozdních endosomů/lysosomů je zprostředkováno sortilinem, je tedy MPR nezávislé.
Lysosomální hexosaminidasa má svůj vlastní aktivátor syntetisovaný nezávisle na prosaposinu. I tento aktivátor může být postižen hereditární mutací. Popis příslušného klinického fenotypu je podán níže.
U některých z enzymů jsou známy proteinové protektivní faktory. Dobrým příkladem může být katepsin A (do lysosomů je cílen prostřednictvím sortilinu). Popis genetického deficitu tohoto proteinu je uveden níže.
Jiný mechanismus, než transport s pomocí MPR, využívají lysosomální membránové proteiny. Jejich specifické cílení do pozdních endosomů/lysosomů je zprostředkováno tyrosinovými a dileucinovými motivy v jejich cytosolicky orientovaných C-koncích a interakcí s celou řadou adaptorových proteinů (AP) na cytosolické straně trans-Golgi membrány. Zároveň opouštějí trans-Golgi cisternu v jiném typu opláštěných váčků (s rozsáhlou specifickou proteinovou výbavou), než jsou ty, které nesou komplexy s MPR. Rezidentní proteiny ELS membrán mohou pozdních endosomů/lysosomů dosáhnout buď přímo nebo nepřímo prostřednictvím cytoplasmatické membrány. Další detaily cílení ELS membránových proteinů přesahují rámec a účel tohoto textu.
Konečnou fází životního cyklu lysosomálních proteinů je proteolytická degradace. Logicky (doposud nebylo přímo experimentálně prokázáno) se nabízí možnost autodegradativní schopnosti lysosomů nejspíše prostřednictvím autodegradace jejich membrán vchlipováním a odškrcováním (podobně jako u multivesikulárního tělíska). V procesu degradace ELS nelze vyloučit i účast MALS (makroautofagie).
Transkripční regulace produkce lysosomálních enzymů byla dlouhou dobu málo prozkoumaná, i když existovala celá řada indicií svědčících pro koordinovanou a společnou expresi celé řady lysosomálních proteinů. Revolučním je v tomto pohledu v nedávné době publikované zjištění, že většina lysosomálních genů podléhá společné transkripční regulaci zprostředkované transkripčním faktorem TFEB (transcription factor EB). S ohledem na tuto skutečnost je pak možné o množině genů kódujících pozdně endosomální/lysosomální proteiny uvažovat jako o jakési transkripčně propojené a koordinovaně exprimované síti, která zásadním způsobem ovlivňuje biogenesi celého ELS, a to jak za fysiologických, tak i patologických stavů. Dobrým přikladem vlivu této koordinované TFEB zprostředkované exprese lysosomálních proteinů mohou zřejmě být situace extrémních sekundárních elevací exprese (hodnoceno nárůstem enzymatické aktivity) celé řady lysosomálních hydrolas u izolovaných deficitů jiné lysosomální hydrolasy.
Zjednodušeně - jedna lysosomální aktivita je deficitní a kompensatorně pak dochází ke koordinovanému nárůstu exprese řady dalších pozdně endosomálních/lysosomálních proteinů (výsledkem je sekundární elevace ostatních nedeficitních hydrolytických aktivit).
Vedle TFEB byly popsány i další způsoby transkripční regulace exprese lysosomálních genů. U hlodavců byla prokázána indukce syntesy β-glukuronidasy androgeny. Dále je známo, že estrogeny snižují sekreci lysosomálních enzymů osteoklasty, zatímco v mléčné žláze indukují produkci katepsinu B (lysosomální proteinasa). U krys byla popsána přímá positivní regulace prostatické kyselé fosfatasy testosteronem na úrovni transkripce.
Lysosomální enzymy mohou být secernovány do extracelulárního prostoru. Jak již bylo uvedeno, určitá a nezanedbatelná frakce lysosomálních enzymů je mechanismem konstitutivní sekrece exportována do extracelulárního prostoru i za normálního stavu (viz výše). Konstitutivní sekrece lysosomálních enzymů je fyziologicky výrazně přítomna v řadě buněčných typů, zejména v mesenchymu. Tento proces může být však také významně zvýšen, zejména u nádorově transformovaných buněk. Lze předpokládat, že rozhodující molekulární alterací v těchto buňkách (nádorových) je snížení syntesy M-6-P značky nezbytné pro cílení enzymu do pozdně endosomálního/lysosomálního systému.
Vedle konstitutivní sekrece mohou být klasické lysosomy exocytovány regulovaně. V některých žlazkách je tento regulovaný typ sekrece těchto enzymů dokonce velmi intensivní (typicky tzv. sekretorická kyselá fosfatasa v prostatě).
Klasickým příkladem buněk s vysokou mírou konstitutivní sekrece lysosomálních enzymů jsou osteoklasty. V tomto buněčném typu sekrece lysosomálních hydrolas dominuje nad jejich cílením do lysosomálního kompartmentu.
Zde je vhodné zmínit genetickou poruchu, postihující jeden z lysosomálních enzymů konstitutivně exocytovaných osteoklasty - katepsin K. Jeho mutace vede k těžké kostní poruše - pyknodysostose, způsobené nerovnováhou mezi degradací a syntesou kostní tkáně, posunutou ve prospěch osteosyntesy.
Za nejrůznějších reaktivních stavů je zvyšována sekrece lysosomálních enzymů aktivovanými makrofágy, které jsou transformovány na tzv. epiteloidní histiocyty, známé ze skupiny tzv. specifických zánětů. Do této kategorie svým způsobem patří i některé střádací buňky, typicky buňky Gaucherovy, známé sekrecí řady cytokinů a obrovských kvant enzymu chitotriosidasy. Sekreční aktivita těchto buněk je používána jako biomarker závažnosti (měření aktivity chitotriosidasy v plasmě) ale tajé efektivity terapie Gaucherovy nemoci (viz níže).
Aktivace určitého typu tkáňově specifických rezidentních makrofágů v rámci lysosomálního střádání, konkrétně mikroglie, je považována za významný CNS specifický patogenetický mechanismus u celé řady lysosomálních onemocnění (viz níže).
Doporučená literatura:T. Braulke, J.S. Bonifacino: Sorting of lysosmal proteins.( BBA 1793:605-614, 2009)
B.A. Schroder et al.: The proteome of lysosomes. (Protemics 10:4053-4076, 2011)
Existují tři základní cesty importu substrátů do pozdních endosomů/lysosomů - endocytosa, autofagie a fagocytosa (Schéma 2). Míra využití těchto tří základních cest může být různá v různých buněčných typech.
Endocytosa (Schéma 3) je obecný termín používaný pro popis procesu internalisace extracelulární tekutiny nebo jiného extracelulárního materiálu invaginací cytoplasmatické membrány. Proces endocytosy zahrnuje následné intracelulární třídění, distribuci a recyklaci internalisovaného materiálu. Pojem endocytosa je z buněčně biologického pohledu velmi široký a zahrnuje celou řadu molekulárních variant a sdružených signalizačních cest. Molekulární mechanismy všech typů endocytosy sledují obecné koncepty vesikulárního transportu sestávají se z následných sekvenčních kroků: (i) membránová invaginace plášťovými proteiny, (ii) uvolnění transportních váčků, (iii) recyklace molekul plášťových proteinů nebo receptorů a (iv) vektoriální transport k akceptorovému kompartmentu a fúze s ním. V tomto textu se omezíme na endocytosu, která představuje dominantní způsob importu extracelulárního materiálu do ELS k degradaci. Tato endocytosa je zprostředkována receptory a je indukována vazbou ligandu na příslušný receptor. Tím je vysoce efektivní. Tento způsob endocytosy je, mimo jiné, biologickým základem substituční enzymové terapie lysosomálních poruch (viz níže).
Po vazbě ligandu na receptor se vytváří endocytický váček, jehož vytváření se zúčastňuje řada proteinů, které též určují jeho další osud. Neznámější je klathrin, určující sferický tvar, a dynamin, jehož funkcí je obtočení a přerušení krčku vzniklého váčku (tuto funkci vykonává i mimo endocytosu). Velká serie dalších proteinů zúčastňujících se na dynamice a regulaci endocytosy je podrobně rozvedena v příslušné literatuře (viz níže)
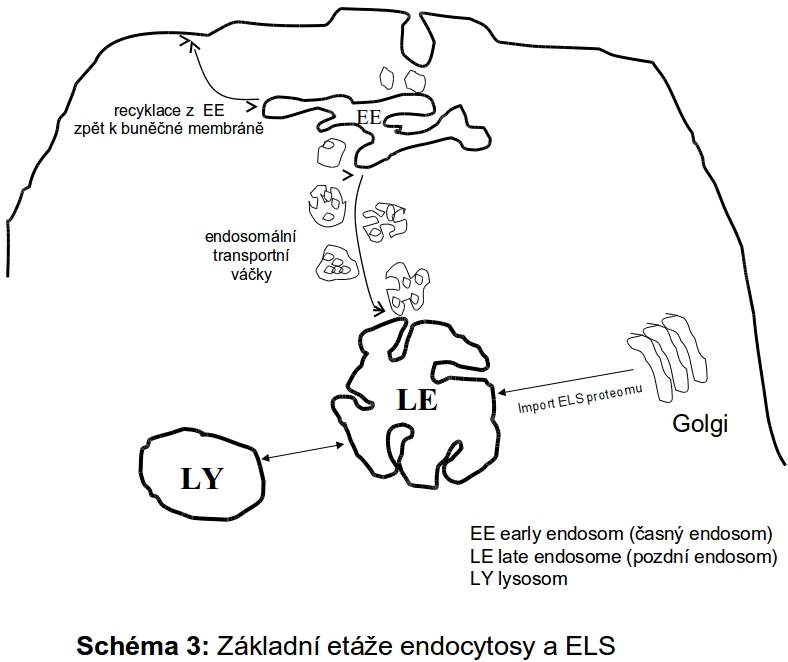
Následná stadia procesu endocytosy jsou charakterizována dvěma základními parametry - molekulárním tříděním v definovaných úrovních endosomální dráhy (časné a pozdní endosomy, viz níže) a progresivním poklesem intraluminálního pH od časných endosomů k lysosomům.
Endocytované molekuly jsou z extracelulárního prostoru invaginací membrány transportovány v drobných opláštěných váčcích do časných endosomů (intraluminální pH 6.0-6.2). Endocytované komplexy ligand-receptor při tomto pH disociují a receptory jsou recyklovány zpět k inserci do cytoplasmatické membrány v recyklujících endosomech. Z časných endosomů endocytovaný materiál postupuje dále ve formě endocytických transportních váčků (endocytotic transport vesicles, ECV) po cytoskeletu do pozdních endosomů, při čemž dochází ke splývání limitujících membrán a obsahů. Plně funkční degradační kompartment (lysosom) pak vzniká "maturací" (jde o dynamický přechod bez jednoznačného morfologického rozhraní) provázenou dalším poklesem luminálního pH. (4.5-5.0). Kyselé pH je kritické pro optimální enzymatickou funkci lysosomálních luminálních hydrolas (viz výše).
Celková organizace endosomální dráhy je založena na principu transientních membránových interakcí a třídících a recyklačních úrovní. Nutno zdůraznit, že není jednoznačně dořešeno, zda se jedná o postupně se transformující (zrající) membránové kompartmenty, či zda dochází pouze k propojení trvalých etáží procesem vesikulárního transportu.
Jak bylo uvedeno, endocytosa je vlastností všech buněk. Velmi výrazná je v hepatocytech, epitelu proximálních kanálků ledvin, folikulárních buňkách štítné žlázy nebo histiocytech. V rámci tohoto textu bude používán termín endocytosa pouze pro proces receptorově podmíněné endocytosy.
Vedle zmíněné klatrin-dependentní endocytosy existuje endocytosa vázaná na systém tzv. kaveol, zprostředkovávajících proces tzv. potocytosy (viz níže). Cílovým určením materiálu internalizovaného tímto typem endocytosy není ELS, ale zejména ER a Golgiho aparát. Tento proces vychází z kaveol (jamek) buněčné membrány, bohatých na protein kaveolin. Jsou v nich koncentrovány i membránové mikrodomény zvané rafty. Tento typ endocytosy je definován jako klathrin nezávislá endocytosa, čímž je odlišná od shora uvedené enydocytosy klathrin-dependentní. Považuje se za východisko transcytosy (viz níže). Touto cestou vstupují do buňky některé ligandy (raft dependentní cesta) nebo viry.
Oproti klathrin dependentní a klathrin independentní endocytose, které jsou selektivní pro určité skupiny ligandů, existuje takzvaná konstitutivní, nespecifická endocytosa vyjádřená ve všech buněčných typech. Jde o endocytosu extracelulární tekutiny, nazývanou též pinocytosou (endocytosa vodné fáze - fluid phase endocytosis). Není závislá na klathrinu
Makropinocytosa je kvantitativně více vyjádřeným procesem pinocytosy, na kterém se účastní i cytoskelet vytvářením klepetovitých výběžků buněčné membrány na povrchu buňky.
Markerem pinocytosy může být křenová peroxidasa, nebo dextran či některá organická barviva (Lucipher Yellow).
Doporučená literatura:G. Scita, P.P. DiFiore: The endocytic matrix. (Nature 463:464-473, 2010)
P. Lajoie, R. Nabi: Lipid rafts, caveolae, and their endocytosis ( Internat.Rev. Cell and Mol.Biol. 282:135-163, 2010)
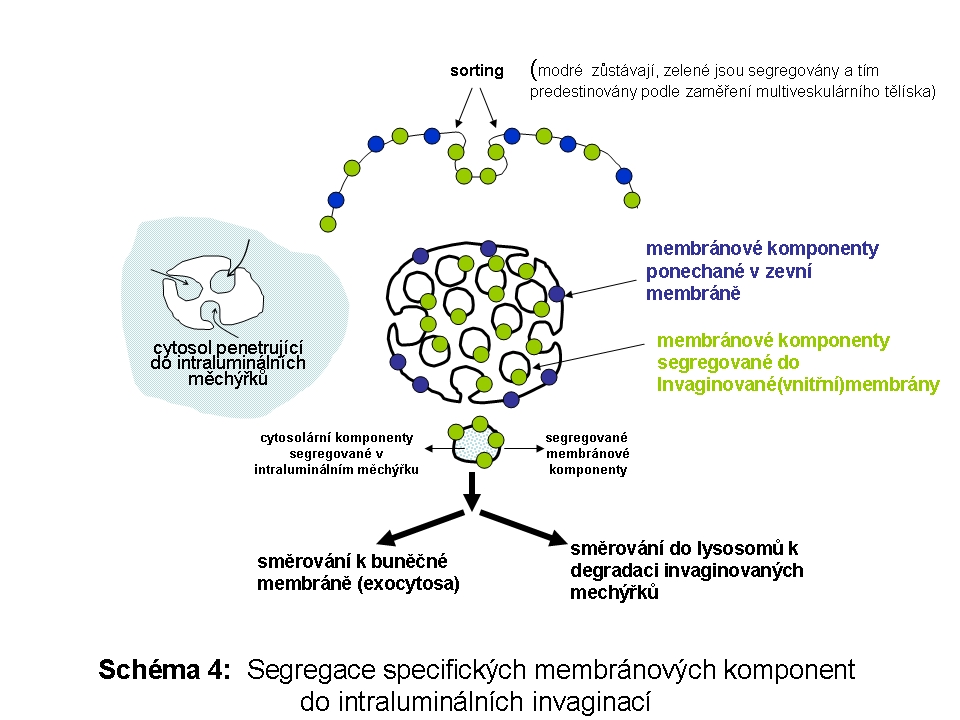
Jde o systém komunikujících vesikul a tubulů, který přijímá endocytotické váčky (Schéma 4). V této části nastává rozpojení ligandy od receptoru vlivem snižujícího se pH uvnitř systému a recyklaci receptoru transportním puchýřkem zpět na buněčnou membránu. V literatuře se lze setkat s názvem CURL (compartment of uncoupling of receptors and ligands). Spojnicí mezi časným endosomem a pozdním endosomem jsou endosomální transportní váčky (endosomal carrier vesicles), pučící z časného endosomu, tzv. multivesikulární tělíska, tvořená tubulárními vchlípeninami zevní membrány do lumen. Ukazuje se, že na tyto váčky lze pohlížet jako na dynamický membránový systém propojení buněčné membrány a časného endosomu s pozdním endosomem. Komponenty určené k inkorporaci do limitující membrány lysosomů jsou lokalizovány v zevní membráně multivesikulárního tělíska, komponenty určené k degradaci jsou (i) ve společném lumen nebo v lumen luminálních invaginací, a (ii) v membránách intraluminálních invaginací. Tyto intraluminální invaginace jsou uvolněny do lumen lysosomu, kde kompletně podléhají degradaci. Komponenty zevní membrány multivesikulárního tělíska jsou včleněny do membrán lysosomálního kompartmentu. Strukturně identické multivesikulární tělísko může být v určitých buňkách určeno k exocytose, zejména v buňkách angažovaných v regulaci imunitní odpovědi. V takovém případě má tato struktura funkční název exosom.
Specifickým způsobem je organizována endocytická dráha v polarisovaných buňkách, jako jsou epitelie (typicky u cylindrických výstélkových epitelů nebo trámčitého epitelu jater) nebo neurony. Tyto buňky mají funkčně stratifikován povrch své cytoplasmatické membrány (u epitelií apikální vs. basolaterální povrch; u neuronů dendrity a neuronální soma vs. axon). Endocytosa z těchto oddělených povrchů (u epitelií těsnými spojkami - "tight junctions"; u neuronů proteinovým aparátem v oblasti tzv. "axon hillock") probíhá prostřednictvím separovaných časných endosomů. Třídící a degradativní integrace pak probíhá na strukturálně hlubších úrovních ELS. U epiteliálních buněk (např. hepatocyty) se tento integrující membránový kompartment nazývá "apical sorting compartment" a zajišťuje specifické třídění i materiálu endocytovaného basolaterálně na apikální membránu (může tak participovat na transcytose, viz níže).
V řadě buněčných typů, ve kterých je exprimován systém organel blízkých lysosomům (viz LRO - lysosome related organelles), představuje časný endosom křižovatku, kde dochází k třídění a cílení do specifického systému granul LRO systému (viz níže).
je vlastní degradační částí systému. Existuje plynulý přechod mezi MVB (Schéma 4) a pozdním endosomem/lysosomem. Je místem nejvyšší koncentrace MPR (viz výše), a tedy cílení vesikulárního transportu lysosomálních enzymů z trans-Golgi zóny. V jeho limitujících membránách jsou specifické komponenty, z nichž je celá řada známá. Jde o transmembránové glykoproteiny s krátkým C koncem situovaným do cytosolu (viz výše) a výraznou glykosylací intraluminálních domén. Této masivní glykosylaci se připisuje ochranný význam před natrávením vlastní limitující membrány lysosomálními hydrolasami. Jak bylo zmíněno jinde v textu, mezi tyto proteiny patří LAMP proteiny 1 a 2 (lysosomal associated membrane proteins). Jejich funkce není plně definována, nicméně LAMP2 proteinu byla přisouzena významná role nejen v procesu fagocytosy ale zejména autofagie, konkrétně makroautofagie, ale i CMA (chaperon mediated autophagy). Vedle LAMP proteinů zahrnuje membránový ELS proteom řádově desítky dalších proteinů. Pokud by měly být některé jmenovány, pak: CD63, LIMP2 nebo membránová H+ ATPasa. Byly objeveny další lysosomální (neenzymové, část z nich je membránová) proteiny, jejichž funkce není známá, ale jejichž mutace vede k neurolysosomálnímu střádání (viz níže).
Tato část lysosomálně endosomálního systému je mnohočetná a pravděpodobně funkčně heterogenní. Strukturálně jde i za normální situace o velmi pleiomorfní systém. Může být představován vakuolami nebo komplexními tubulocisternálními strukturami blízkými trans- Golgi zoně. V této části endosomálního systému dochází ke skutečné funkční integraci transportu substrátů a jejich degradujících enzymů (viz výše).
Konsensuálně posledním stadiem zrání endosomu je lysosom (tzv. "densní lysosom"), který neobsahuje MPR. Má tedy pouze residuální sadu lysosomálních enzymů a integrální membránové specifické proteiny v limitující membráně. Je považován za terminální část, v určitém smyslu residuální, často obsahující lipofuscin (viz dále). Nicméně podle řady studií je komunikace na této strukturální úrovni s pozdními endosomy velmi intenzivní, takže jejich ireversibilní terminální povaha je sporná.
Dřívější členění lysosomálního systému je opuštěno. Původní tzv. primární lysosomy odpovídají nejspíše současným transportním váčkům transportujícím lysosomální enzymy do pozdního endosomu. Původnímu pojmu lysosom by měly odpovídat dva oddíly dnes popisovaného endosomálního systému, a to pozdní endosom a lysosom (pozdní endosom/lysosom, viz použití pojmů).
se odlišuje od endocytosy tím, že (i) neprobíhá konstitucionálně, (ii) neúčastí klathrinu, (iii) závislostí na aktinu, který umožňuje dynamiku obchvácení partikule výběžky cytoplasmy, (iv) na zvětšování membrány fagosomu se podílí i vesikulární transport z endoplasmatického retikula, což významně šetří buněčnou membránu. Proces je z větší části závislý na receptorech (Fc receptory u histiocytů), ale v řadě případů není mechanismus interakce fagocytované partikule s buněčnou membránou fagocytu známý.
Prvou fází fagocytosy je vznik fagosomu, jehož membrána je přímo odvozena od buněčné membrány. Podmínkou úspěšné degradace fagocytovaného materiálu je transformace fagosomu na fagolysosom. Tím je zajištěn příjem lysosomálních enzymů. Je to podmíněno kontaktem s pozdním endosomem, při kterém nastává současně s přenosem lysosomálních enzymů i přenos membránových komponent. V tomto kontaktu hraje významnou roli posun obou kompartmentů (nebo jejich tubulárních výběžků) po mikrotubulech. Tím je transformace na fagolysosom dokončena. Blok této transformace má za následek poruchu digesce fagocytovaného obsahu. K takové situaci dochází u některých bakteriálních infekcí, což má negativní dopad na imunitní odpověď. Blíže je o zásadním významu efektivní fagocytosy pro imunitu pojednáno níže v oddílu "Role ELS v imunitě". Podobně jako v případě fúze autofagosomů s pozdními endosomy je i v případě fúzí fagosomů s pozdními endosomy/lysosomy jedním z kritických proteinů LAMP2. Ztráta těchto funkcí je podkladem hereditárního deficitu LAMP2 - Danonovy nemoci (viz níže).
Fagocytosa je vlastností všech buněk. Nejvýraznější je v případě histiocytů (makrofágů), pro které se používá název profesionální fagocyty. Tyto buňky normálně zajišťují kontinuální likvidaci terminálních populací hematologických klonů, za patologických situací ireversibilně poškozených buněk. Jejich lysosomální systém je permanentně zatěžován. Za některých situací jsou vystaveny nadměrné zátěži (viz níže - získané poruchy ELS). Stupeň fagocytární aktivity histiocytů a pohotovost k digesci jsou do značné míry ovlivněny lymfokiny produkovanými T buňkami (např. makrofágy stimulující faktor). Empirickým markem makrofágů je molekula značená jako CD68, proti níž existují různé monoklonální protilátky dostupné komerčně (viz níže - detekce lysosomů)
Fagocytosa u jiných buněčných typů mimo makrofágy je vyjádřena zejména v procesu apoptosy (viz samostatná kapitola buněčná smrt). Apoptotická buňka je fagocytována okolními buňkami nezávisle na jejich buněčném typu. Fagocytosa realisovaná granulocyty a makrofágy je spojena s výraznou a velmi důležitou metabolickou reakcí tzv. oxidativním vzplanutím, při kterém dochází, vedle jiných, také k produkci volných radikálů. Jde o aktivitu NADPH oxidasy, multipodjednotkového enzymu, který se asembluje do aktivního stavu v průběhu fagocytosy. Výraznou schopnost fagocytosy má pigmentový epitel sítnice (kontinuální fagocytosa terminálních částí fotoreceptorů).Výraznou fagocytární schopnost mají i keratinocyty epidermis, které kontinuálně fagocytují melanosomy syntetisované v melanocytech. Fagocytosu erytrocytů lze například pozorovat i v tkáňové kultuře fibroblastů (Obr. 1).
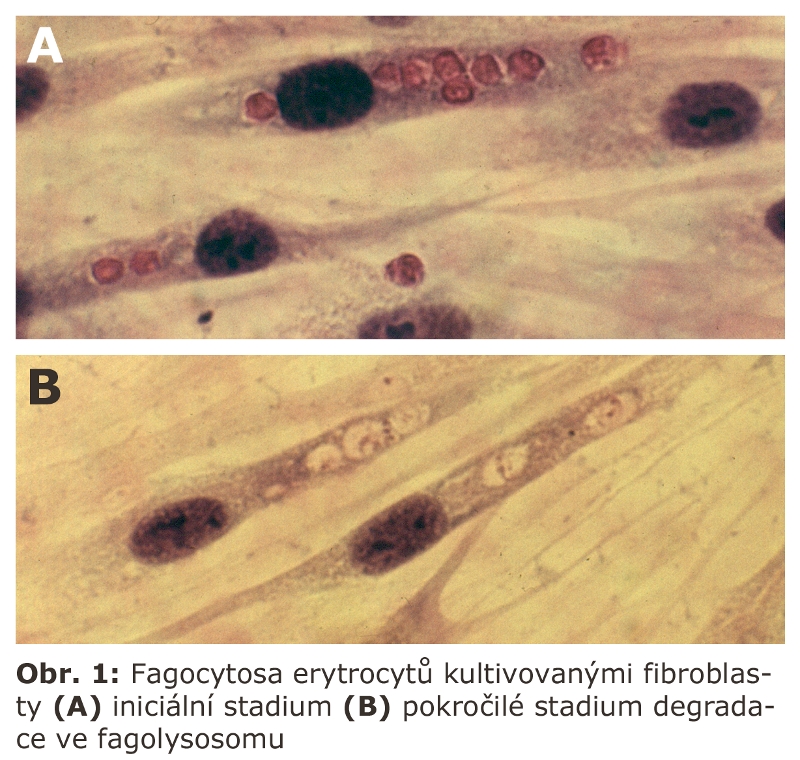
Jde především o transcytosu, která je výrazem vysoké polarity buňky. Jde o mechanismus, kterým se látky transportují přes buňku uzavřené v transportních vesikulech na opačný pól buňky, kde je obsah těchto transportních vesikul uvolněn do extracelulárního prostoru. Proces je iniciálně odvozen od časných endosomů, dále se však osamostatňuje.
Tímto způsobem například probíhá přenos imunoglobulínů (Ig) z basolaterální části střevního nebo respiračního epitelu k povrchu apikálnímu a na povrch sliznice. V hepatocytu se tímto způsobem přenáší řada látek (včetně Ig) ze sinusového pólu (basolaterální membrána) na žlučový pól (apikální membrána) a do žlučového systému (viz výše). Výrazný je tento proces také v cévním endotelu a v placentě při přenosu látek přes trofoblast.
Potocytosa je proces regulovaného vniku malých molekul přes plasmatickou membránu na specializovaných místech buněčného povrchu, organisovaných do tzv. jamek (caveolae). Na cytoplasmatické straně membrány je koncentrován specifický cytoplasmatický protein caveolin, jehož funkce není doposud objasněna. Potocytosa může hrát důležitou roli v přenosu látek do extralysosomálních oddílů buňky nebo přes buňku, zejména v kapilárách (viz výše).
Velmi podobná fagocytose je peripolesa a emperipolesa. Jde o proces vzájemného vchlipování až kompletní penetrace jedné buňky do druhé. Tento fenomén je pozorovatelný jak za normy tak za patologických stavů. Na rozdíl od fagocytosy při něm nedochází k destrukci penetrující buňky buňkou recipientní. Za normálního stavu se vyskytuje ve dřeni kostní, kde různé krevní elementy invadují do megakaryocytů. V GIT invadují T-lymfocyty do epitelií a participují na mechanismech slizniční imunity. Je pozoruhodné, že za patologických stavů je recipientní buňkou modifikovaný histiocyt obsahující velmi četné lymfocyty, plasmocyty a granulocyty (sinusová histiocytosa s masivní lymfadenopatií - sy. Rosai-Dorfman). Membrána vakuoly je nejspíše identická s buněčnou membránou hostitelské buňky a dále se netransformuje. Význam tohoto děje není jasný.
Autofagie je proces vlastní všem buněčným typům lidského těla. Jedná se o složitý systém signalizačních a exekutivních drah, který buňkám dovoluje efektivní eliminaci vlastní cytoplasmy (proteinů, biologických membrán či celých organel). Autofagie probíhá jako konstitutivní (stále probíhající) proces s malou intensitou nebo jako indukovatelný mechanismus spouštěný stresovými podmínkami (např. buněčným hladověním). V rámci autofagocytosy dochází buď k likvidaci větších poškozených cytoplasmatických fokusů (tzv. fokální cytoplasmatická degradace). Takto neselektivní autofagie má svá definovaná stadia a regulativní mechanismy. Jedná se o významný ubikviterní proces, kterým se odstraňují nejen poškozené, ale i nadbytečné části cytoplasmy.
Proces autofagie je aktivován při přechodu hypertrofie v eutrofii nebo v atrofii a v opačném případě je utlumen. Při hladovění je autofagocytosa velmi aktivní a slouží jako mobilizovatelný záložní zdroj energie. Za těchto situací dosahuje autofagocytosa vysokých hodnot zejména v hepatocytech, ve kterých byl celý proces studován i kvantitativně s tím, že v průměru 4% sledovaných cytosolických enzymových aktivit za 1 hodinu přecházelo do korpuskulární lysosomální frakce (tedy bylo autofagocytováno). Předpokládá se, že u experimentálních zvířat je 20-30% celkového hepatocytárního proteinu degradováno během 24 hodin hladovění. Jedna z forem autofagie (MALS, viz níže) se pokládá za exekutivní dráhu programované buněčné smrti typu II (viz samostatná kapitola Buněčná smrt).
Proces hepatocytární autofagocytosy je ovlivnitelný řadou látek. Inhibičně působí aminokyseliny, zejména v kombinaci s insulinem, dále epinefrin, puriny (takřka kompletně inhibuje 3 methyladenin) a cyklické nukleotidy. V experimentu se autofagocytosa indukuje vinblastinem nebo perfusí jater mediem chudým na aminokyseliny s glukagonem.
Autofagie je evolučně konzervovaná signalizační a exekutivní buněčná dráha, která je integrální součástí sítě mechanismů proteostasy (homeostasa proteinů) společně s UPS (viz výše). Fysiologické role autofagie v degradaci proteinů nebo ontogenesi, stejně jako implikace těchto drah pro lidskou patologii včetně variantních programů buněčné smrti (typ II programované buněčné smrti, viz samostatná kapitola Buněčná smrt) jsou v posledních několika letech intensivně studovány. Autofagie se například zdá být jedním z hlavních faktorů mobilisace glykogenu u novorozenců. Je velmi pravděpodné, že v nejbližší době budou, pokud již nebyly, odhaleny další zásadní role autofagie a přidružených drah v patogenesi lidských onemocnění. Vedle lysosomálních střádacích onemocnění (viz níže) to zajisté budou nejrůznější neurodegenerativní stavy nebo nádorová onemocnění.
V současnosti jsou rozlišovány tři základní molekulární varianty autofagie (Schéma 5): makroautofagie (synonymně makroautofagicko lysosomální systém - MALS - macroautophagy/lysosomal system), mikroautofagie a chaperony zprostředkovaná autofagie (CMA - chaperone - mediated autophagy).
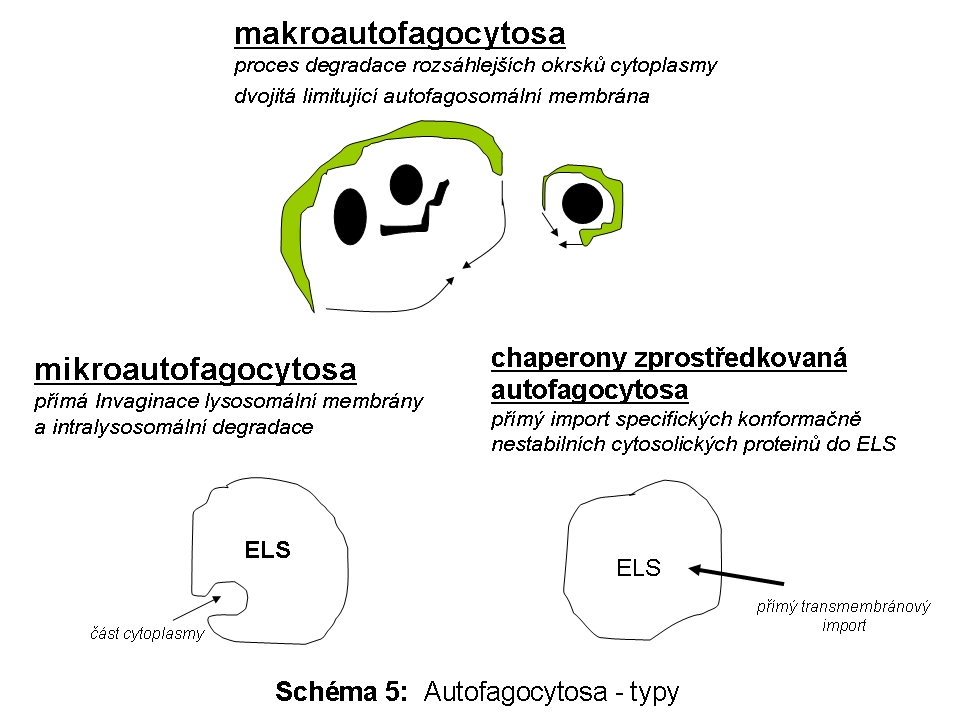
označuje proces, při kterém jsou poškozené organely nebo úseky cytoplasmy ohraničeny autofagosomální membránou (typickým "substrátem" makroautofagie jsou zejména: mitochondrie, peroxisomy či sekreční granula).
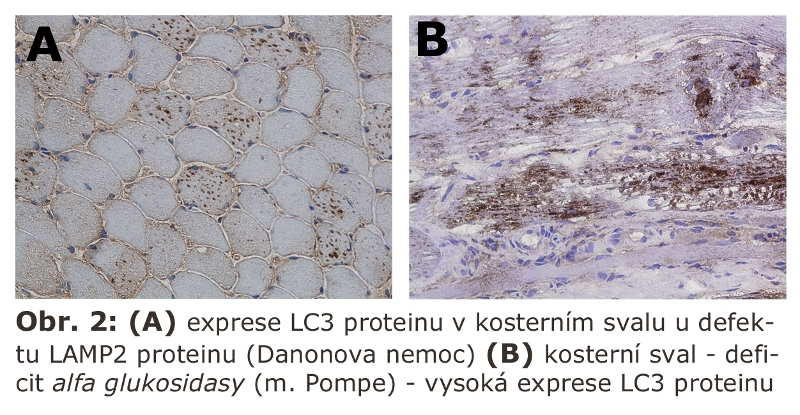
Přesný původ dvojité autofagosomální membrány (de novo syntesa nebo pre-existující mobilizovatelné, ale nefúzované vesikuly vs. derivát preexistujících membrán, jako třeba cisterna hladkého endoplasmatického retikula či Golgiho aparátu) není stále definitivně objasněn. Jasné je, že autofagická sekvestrace je realisována dvojitou membránou. Ohraničením části cytoplasmy s nejrůznějšími strukturami vzniká uzavřený autofagosom, který neobsahuje ani lysosomální enzymy, ani membránové komponenty lysosomů. Proteom autofagosomální membrány a autofagosomů byl v savčích buňkách definován, předpokládá se nezávislý přísun z Golgiho aparátu. Regulační proteinové dráhy, které participují na vzniku a propagaci autofagosomální membrány, jsou v současné době velice intensivně studovány. V oblasti výzkumu procesů spojovaných s makroautofagií sehrál a sehrává významnou roli kvasinkový eukaryotický model, ve kterém byla nalezena celá řada genů/proteinů (Atg - autophagy related genes) zodpovědných za proces makroautofagie. Znalost genů u kvasinky dovolila jednodušší a rychlejší identifikaci savčích (včetně lidských) ortologů. Má-li být jmenován jeden protein základním způsobem spojený se vznikem autofagosomální membrány, pak je to microtubule-associated protein light chain 3 (LC3B). LC3B je za normálních okolností cytosolický protein. V tomto stavu je označován jako LC3B-I. V případě indukce autofagie a zahájení formování makroautofagosomální membrány je tato forma (LC3B-I) sekvenčně proteolyticky štěpena a v konečné fázi konjugována s fosfatidyletanolaminem. Tato konjugovaná forma, jinak označovaná jako LC3B-II (Obr. 2) je, znovu za pomoci jiných proteinů, integrována do rostoucí autofagosomální membrány (do obou listů zevního i vnitřního). Existují důkazy, že je to právě LC3B-II protein, který zajišťuje hemifúzi autofagosomální membrány (viz následující odstavec).
V dalším průběhu autofagocytosy se autofagosom transformuje v autofagolysosom prostřednictvím membránových fúzí s preexistujícími pozdními endosomy/lysosomy. Touto transformací a fúzí s existujícími pozdními endosomy/lysosomy je zajištěna schopnost degradace sekvestrovaných částí cytoplasmy (cytosolu i organel). Prostředníkem těchto membránových fúzí je, mimo celé řady jiných proteinů, i molekula LAMP2, jejíž cytosolická část zároveň zajišťuje interakci s mikrotubulárním cytoskeletem, a tímto způsobem přispívá k intracelulární prostorové distribuci autofagosomů a lysosomů (hereditární deficit LAMP2 viz níže).
Po splynutí autofagosomů s pozdními endosomy/lysosomy a degradaci jejich obsahu jsou produkty hydrolysy recyklovány kombinací pasivní difuse a specifického transmembránového transportu a mohou tak být znovu buňkou využity.
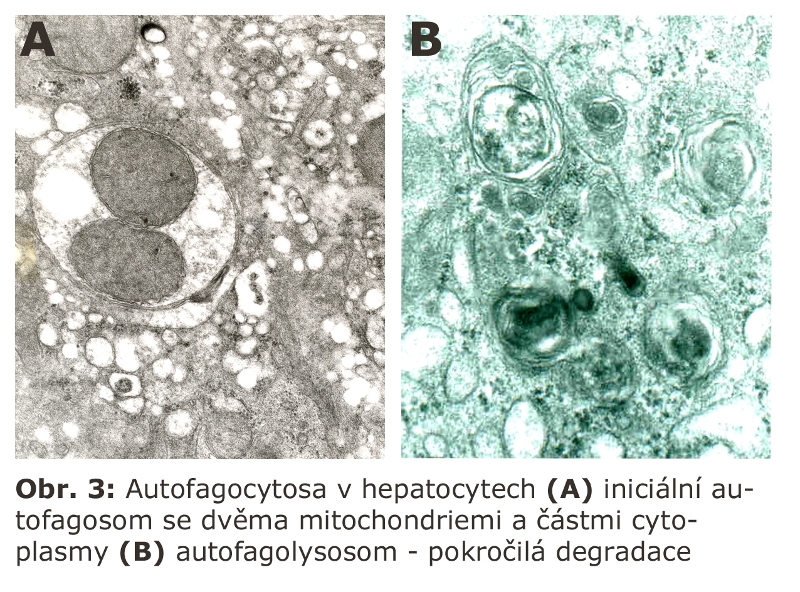
Demonstrace iniciální a pokročilé fáze autofagocytosy v hepatocytu (Obr. 3).
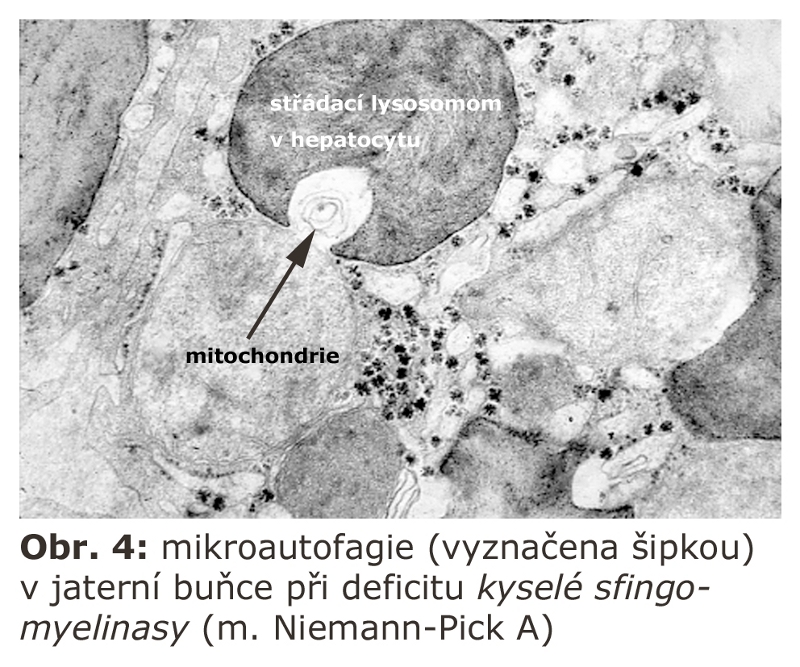
je na rozdíl od MALS mechanismus, při kterém lysosomální membrána přímo obklopí část cytoplasmy nebo malé organely, jako jsou například peroxisomy (pexofagie), vchlipováním lysosomální membrány. Krček takových výchlipek je odškrcen a celá invaginovaná struktura je degradována (podobně jako u multivesikulárních tělísek). Lysosom se tak sám může projevovat jako agresor proti některým organelám nebo částem cytosolu, které do sebe využitím mechanismu mikroautofagie inkorporuje (Obr. 4).
(CMA) je selektivní formou intralysosomální degradace specifické skupiny cytosolických proteinů obsahujících KFERQ motivy v primárních amino kyselinových sekvencích. Pokud takový cytosolický protein v průběhu své existence získá konformačně nestabilní formu, může být vázán cytosolickým a následně lysosomálním chaperonem (hsc70 and ly-hsc70) a prostřednictvím LAMP2A (sestřihová varianta LAMP2) v lysosomální membráně translokován do lumen lysosomu, kde je degradován. Podobně jako MALS i CMA je aktivovatelná stresovými podmínkami, jako jsou například hladovění, oxidativní stres nebo exposice toxickým látkám.
ELS je zapojen do systému intracelulárního vesikulárního transportu, který zajišťuje specifický pohyb biologického materiálu ve vazbě na membránové transportní váčky či membránové kompartmenty obecně. ELS extensivně komunikuje nejen v rámci membránových kompartmentů sobě vlastních (všechny etáže endocytické dráhy, fagosomy, autofagosomy), ale i "navenek" s Golgiho aparátem, ER nebo cytoplasmatickou membránou. To je jedna z velmi výrazných vlastnostní ELS, která svědčí o jeho značné dynamice. Prostřednictvím těchto kontaktů ELS ovlivňuje i intracelulární procesy s primární funkcí ELS nesouvisející, jako je třeba signalisace probíhající na cytoplasmatické membráně nebo v časných oddílech endocytosy.
Dynamika, směrování a specifická interakce jednotlivých částí ELS a dalších membránových kompartmentů se děje za pomoci škály membránových proteinů z kategorie (SNARE). Tyto proteiny jsou specifickými molekulárními značkami intracelulárních membránových kompartmentů a mají schopnost po aktivaci (většinou Rab proteiny) facilitovat fúze lipidních dvouvrstev (včetně jejich proteinové výbavy) původně oddělujících separované membránové kompartmenty. Výsledkem je sdílení luminálního a membránového obsahu. Zároveň tyto proteiny odpovídají za kontakt s cytoskeletem (konkrétně mikrotubuly) prostřednictvím proteinů typu mechanoenzymů z rodiny kinesinů, dyneinů a asociovaných proteinů. V nedávné době se ukázalo, že distribuce a správná funkce zprostředkovatele membránových fúzí je v případě SNARE proteinů závislá na celkovém obsahu cholesterolu v membránách. Je jasné, že celá řada lysosomálních střádacích onemocnění (viz níže) přímo vede k poruše celkového obsahu a změně distribuce nejen cholesterolu, ale i dalších membránových lipidů. S ohledem na tuto skutečnost pak nepřekvapí, že jedním z důsledků lysosomálního střádání je porucha fúzní dynamiky ELS membrán.
Kontakty membránových struktur v rámci ELS se mohou dít buď jako pohyb drobných sférických váčků, nebo (typicky v rámci pozdně endosomálního kompartmentu) jako extrémně rychlá reorganisace membrány do podoby tenkých tubulárních výběžků, které transientně kontaktují jiné membránové kompartmenty (např. cytoplasmatickou membránu). Tímto způsobem je distribuován v rámci pozdně endosomálního/lysosomálního kompartmentu třeba NPC1 protein (viz níže). V této souvislosti je také stále diskutována (zdá se být velmi pravděpodobná) existence membránových mikrodomén v rámci ELS. Tyto by pak mohly zajišťovat nejen vnitřní signalizaci v ELS systému, ale také by mohly zajistit efektivnější způsob sdílení membránových úseků mezi jednotlivými složkami ELS navzájem, nebo ELS a ostatními cytoplasmatickými membránovými kompartmenty.
Zajímavou otázkou zůstává vnitřní organizace ELS při nutnosti zajistit koordinovanou a specifickou distribuci substrátů a příslušných hydrolytických enzymů u sekvenčních vícekrokových degradačních kaskád (typicky degradace glykosaminů u skupiny mukopolysacharidos nebo degradace glykolipidů). Na příkladu degradace glykosaminoglykanů byla demonstrována určitá substrátová exklusivita částí ELS, která nutně musí být provázena odpovídající diferencovanou přítomností jednotlivých hydrolas v rámci ELS. Pro zjednodušení, lze si představit, že ne všechny úseky ELS jsou schopny degradovat díky své residentní enzymatické výbavě všechny typy substrátů. Jak je tento typ třídění substrátů a jejich hydrolas realisován (podle dostupných informací v omezené míře existuje) není jasné.
Podobnou stratifikaci jako v případě zmíněného enzym/substrátového třídění lze očekávat i v míře M-6-P deglykosylace ELS proteinů prostřednictvím tartarát resistentní fosfatasy (ACP5, viz výše). Funkční dopady tohoto fenomenu nejsou plně objasněny.
Zvláštní specifika má ELS systém a jeho vnitřní organizace v polarisovaných buňkách (viz výše). Například v neuronech, ve kterých je ELS systém polarisován jak do dendritů, tak do axonů (oddělené endocytující povrchy cytoplasmatické membrány) a spolupodílí se na zprostředkování interneuronální komunikace. Hlubší kompartmenty ELS jsou lokalisovány perinukleárně, zatímco axon a dendrity obsahují ranější fáze endocytosy. Hezkým příkladem může být organisace endocytické dráhy v axonu, kdy aktivním místem vstupu substrátů je synaptická štěrbina, a transport všech typů endosomů je následně axonem retrográdní. Po délce axonu směrem k neuronálnímu tělu je pak jasně patrný postupný pokles pH v endocytárním kompartmentu. Podobná specifika má kupříkladu axonální autofagocytosa, která probíhá s maximální intensitou v oblasti synaptické axonální terminály (synaptic button) a pohyb autofagosomů je potom retrográdní, jelikož je tímto způsobem zajištěna jejich perinukleární fúze s pozdními endosomy/lysosomy.
Podobný mechanismus endosomálního třídění byl již výše několikrát zmíněn u polarisovaných epitelií (typicky hepatocytů).
V naprosté většině případů je možné si myslet, že lysosomální substrátová degradace je kompletní, a to i u heterozygotů lysosomálních enzymopatií, u kterých je poloviční aktivita příslušné hydrolasy (klinicky jsou tito lidé zdraví). Lysosomální membrána je nepropustná pro makromolekuly a oligomery. Pro monomery se dlouho předpokládala pouhá nespecifická difuse, ale nepochybně existují transportní mechanismy. Lysosomální membránové transportéry byly popsány pro sialovou kyselinu (obecně pravděpodobně pro organické aniony) a pro cystin.
V nedávné době byl popsán mechanismus, jakým je sdílen intralysosomální neesterifikovaný cholesterol mezi dvěma pozdně endosomálními/lysosomálními proteiny, z nichž jeden je intraluminální solubilní (NPC2) a druhý je pevně kotven do lysosomální limitující membrány (NPC1). Zároveň byl navržen model, jak toto sdílení cholesterolu mezi NPC1 a 2 vede k jeho translokaci přes limitující membránu pozdních endosomů/lysosomů. Mutace v jednom z těchto dvou proteinů se vzájemně komplementují v klinickém rozvoji Niemann-Pickovy choroby typ C (viz níže).
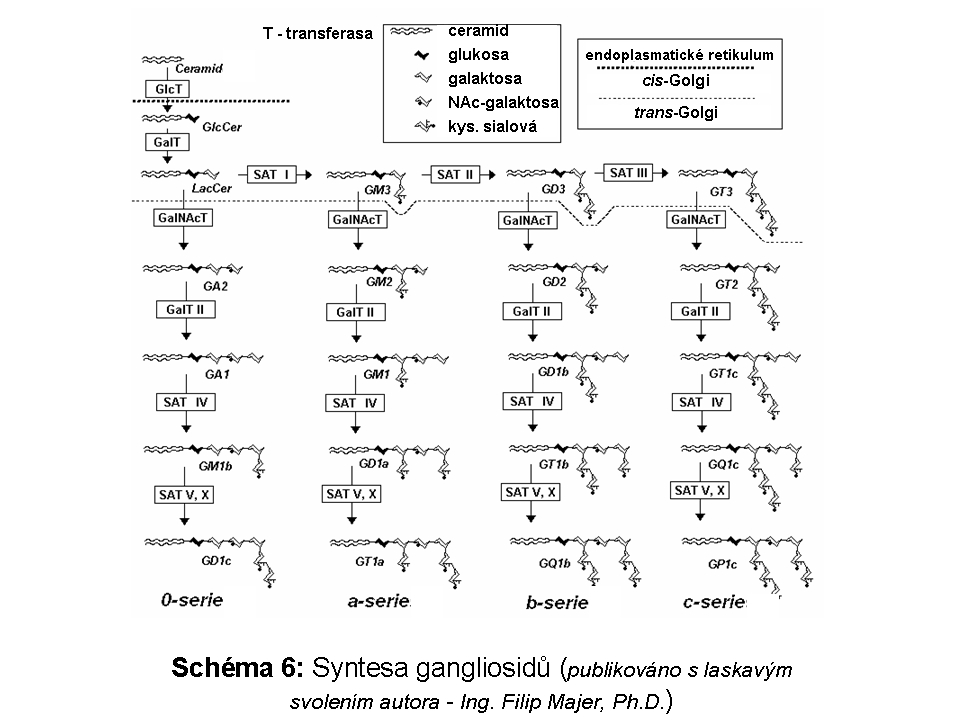
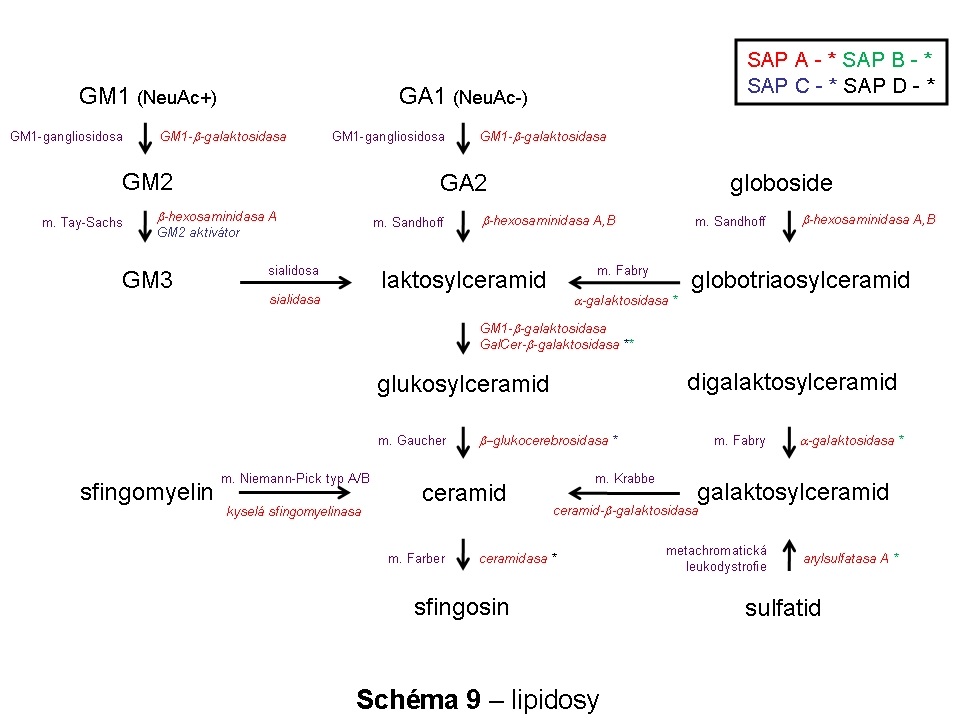
Složitost distribuce a pohybu lipidů v rámci membrán ELS a sdružených kompartmentů (zejména Golgiho aparátu a cytoplasmatické membrány) lze demonstrovat na příkladu gangliosidů. Na tomto příkladu lze také dokumentovat, že ne vždy nutně dochází v ELS ke kompletní degradaci na základní stavební konstituenty a recyklovány mohou být i komplexnější formy. Gangliosidy jsou zčásti syntetisovány (Schéma 6) a finálně upravovány v Golgiho aparátu (konkrétně trans-Golgi) odkud se exocytosou dostávají do cytoplasmatické membrány, kde se mohou stát významnou komponentou mikrodomén plasmatické membrány (membránové rafty), které sehrávají kritickou úlohu v celé řadě signalisačních drah. Z cytoplasmatické membrány jsou endocytovány a dospějí až do degradačních etáží ELS, kde jsou degradovány na monosialogangliosidy (GM1) a následně na GM2 (katalysováno β-galaktosidasou) a GM3 (katalysováno β-hexosaminidasou), tyto jsou pak dále degradovány na neutrální glykosfingolipidy laktosylceramid a glukosylceramid. Část monosialogangliosidů je však z ELS odkloněna do Golgiho aparátu k reglykosylaci, zatímco zbytková frakce je kompletně degradována (Schéma 7 a 9).
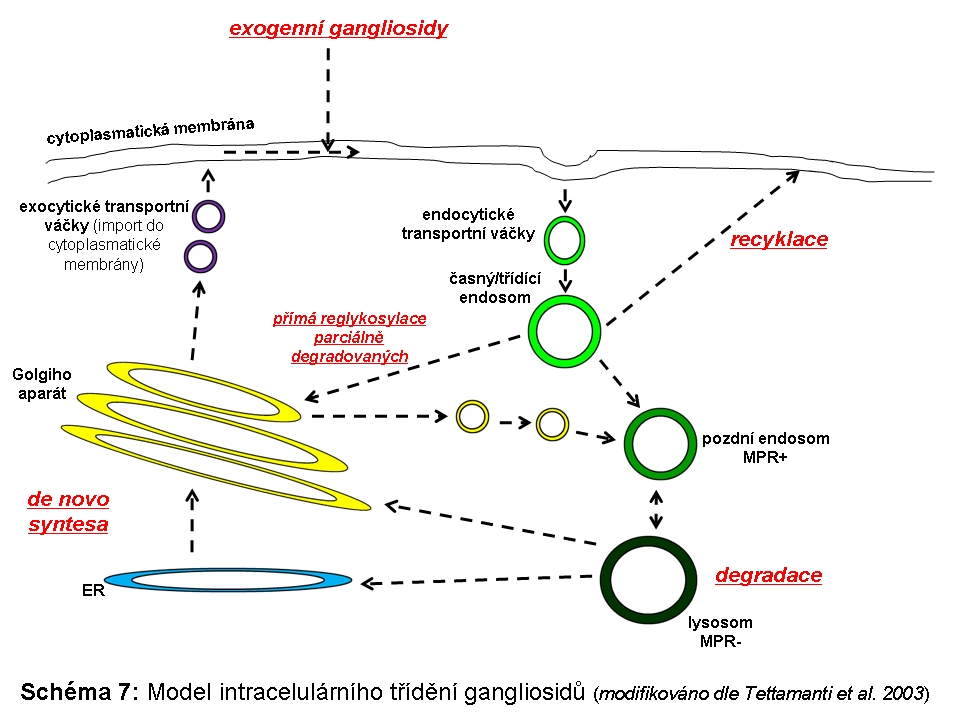
Zásadní pro recyklační funkci ELS, ale i patologii lysosomálních střádacích onemocnění (viz níže) je, zda se podobný mechanismus popsaný u gangliosidů může týkat i jiných recyklovaných substrátů resp. jejich degradačních produktů (např. cholesterol nebo glykosaminoglykany). Podstatná je totiž otázka, jakým způsobem je regulován poměr substrátů určených k degradaci a těch, určených k resyntese, na podkladě inkompletně degradovaných forem.
Fysiologická exocytosa z úrovně pozdního endosomu je přítomna v buňkách exprimujících antigeny pomocí MHC II a CD1. Jde o skupinu antigen presentujících buněk, mezi které patří makrofágy, dendritické buňky a některé další buněčné typy. V těchto buňkách je protein určený k imunitní presentaci regulovaně a pouze zčásti v ELS degradován a jeho fragmenty jsou ve spojení s MHC II proteiny exprimovány exocytosou na buněčném povrchu pro další kontakt s imunokompetentními buňkami (pomocné CD4+ T-lymfocyty). Podobná je situace i u lipidních antigenů. O obou situacích podrobněji viz níže - Role ELS v imunitě).
Experimentálně byla prokázána exocytosa lysosomálního kompartmentu z úrovně pozdního endosomu i nezávisle na buněčném typu. Podmínkou byla přítomnost lysosomálního synaptotagminu VII. Pomocí této lysosomální exocytosy dochází zřejmě k akutní reparaci defektů buněčné membrány. Vyřazením synaptotagminu VII v myším modelu došlo k rozvoji výrazně patologického fenotypu charakterizovaného myositidou s výraznou fibrosou, masivní fibrosou dermis a k indukci tvorby antinukleárních protilátek.
Další ukázkou schopnosti membrán ELS doplňovat cytoplasmatickou membránu může být proces cytokinese, při kterém je nutné ve velmi krátké době významně navýšit celkovou plochu cytoplasmatické membrány. Při konstituci povrchových membrán dceřinných buněk je toto navýšení akcelerováno právě díky koordinované fúzi membrán ELS s cytoplasmatickou membránou.
Obecně lze říci, že známkou exocytosy ELS kompartmentu je přítomnost jeho membránových komponent na buněčné membráně, což lze využívat i diagnosticky (celá řada aplikací průtokové cytometrie).
Do této kategorie lze řadit situace, kdy dochází k reversibilnímu přetížení lysosomálního systému přechodnou zvýšenou náloží substrátů určených k degradaci v ELS. Výsledkem je zvýšená degradace těchto látek v jejich lysosomech (s normální enzymovou výbavou), a z toho plynoucí zvýšení produktů lysosomální degradace, které jsou následně transportovány přes lysosomální membránu do cytosolu či jiných buněčných membránových kompartmentů.
Zmíněny budou základní skupiny. Jde o situace, za kterých jsou buňky, nejen makrofágy, vystaveny látkám v extracelulární tekutině, které aktivují endocytosu (receptorem zprostředkovanou), nebo využívají pinocytosy a jsou tak transportovány do ELS k degradaci. Proces může být, podle povahy, velmi intensivní a může imitovat geneticky podmíněnou lysosomální poruchu. Někdy jde o reakci na intravenosně aplikovanou látku. Pro tyto procesy lze použít termín AFCS (acquired foam cell syndrome), jinak řečeno získaný (sekundární) syndrom pěnitých buněk.
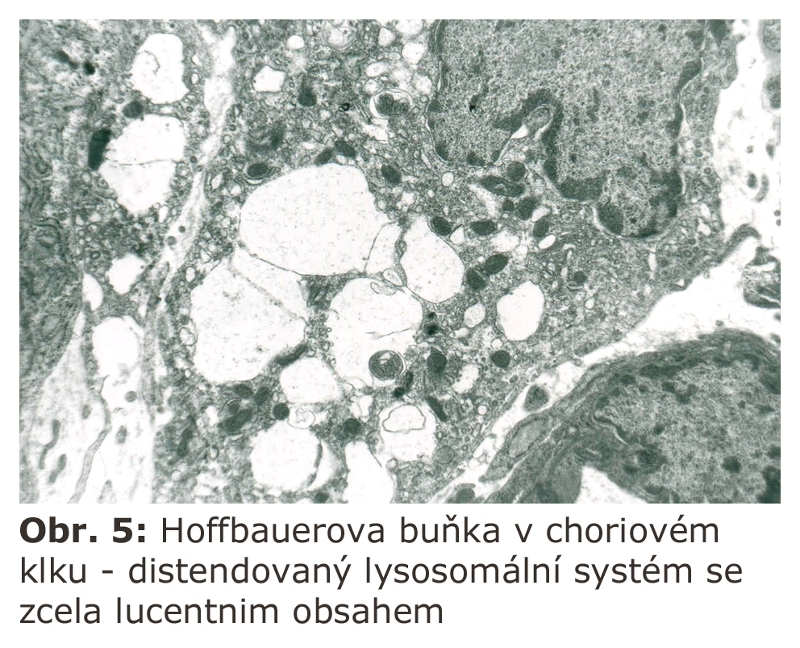
Poznámka. Je všeobecně známo, že existuje jeden typ makrofágu, který má permanentně aktivovaný (a distendovaný) lysosomální systém, aniž by šlo o fagocytosu. Jsou to Hoffbauerovy buňky stromatu choriových klků (Obr. 5). Toto musí být bráno na zřetel při event. elektronoptickém vyšetřování choriových klků pro podezření z lysosomální enzymopatie. Pro tento zvláštní stav lysosomálního systému Hoffbauerových buněk není doposud vysvětlení.
Klasickým příkladem je tzv. osmotická nefrosa (nemusí jít o manifestaci pouze v ledvinách ale i generalisovaně), v obecně biologické terminologii osmotická expanse lysosomů. Jde o stav přetížení renálních (i mimorenálních) lysosomů aplikací di- a pravděpodobně i oligosacharidů. Experimentálně se používá k vyvolání osmotické nefrosy hypertonické sacharosy. Podmínkou je průnik jejího většího množství mechanismem pinocytosy do lysosomů s jejich následnou osmotickou expansí. V lidské patologii je tento obraz běžný v renálních proximálních kanálcích po intravenosní aplikaci většího množství hypertonické sacharosy. Může se manifestovat i extrarenálně (Obr. 6). Podstatou tohoto jevu je relativně vysoká neprůchodnost buněčných membrán pro sacharosu a nemožnost jejího štěpení lysosomálními enzymy. Sacharosa je neredukující disacharid (beta-D-fruktofuranosyl-alfa-D glukopyranosa) štěpený sacharasou/isomaltasou, která funguje v cytoplasmatické membráně kartáčového lemu enterocytů. K podobné situaci dochází i u jiných disacharidů (trehalosa, cellobiosa či turanosa). Přísun příslušné disacharidasy je prevencí této osmotické distense lysosomů. Osmotickou expansi lysosomů lze indukovat sacharosou v jakémkoliv buněčném typu schopném endocytosy. Využívá se proto v experimentech na tkáňových kulturách.
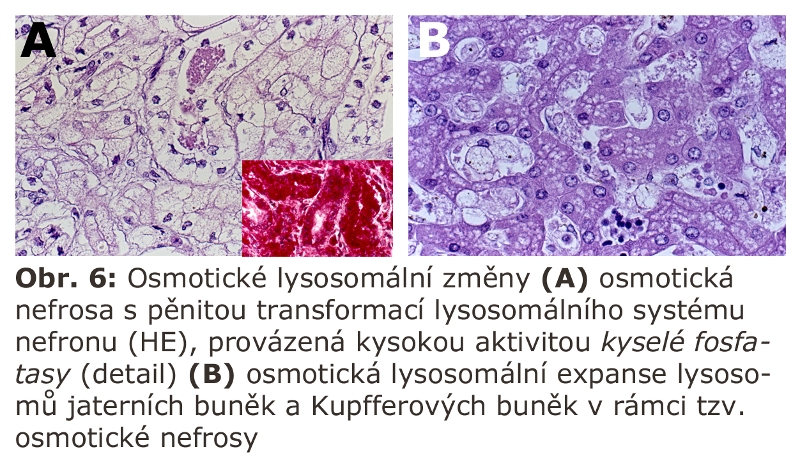
Analogickou situací jsou nálezy při infusi dextranu, který, zřejmě v závislosti na molekulové hmotnosti, může být endocytován řadou buněk, zejména makrofágů.
Dalším klasickým příkladem je tzv. hyalinní zkapenkovatění ledviny (Obr. 7), což je proces omezený na ledviny, způsobený přetížením lysosomálního aparátu proximálních tubulů ledvin při glomerulárních poruchách vedoucích k proteinurii. Nadměrné množství proteinů v primární moči se dostává endocytosou do lysosomálního systému epiteliálních buněk proximálních stočených kanálků prostřednictvím endocytosy zprostředkované polyproteinovými receptory megalinem a cubilinem. Lysosomální aparát těchto buněk je přeplňován a hydrolytická degradace substrátů (v tomto případě proteinů) tak zaostává za přísunem.
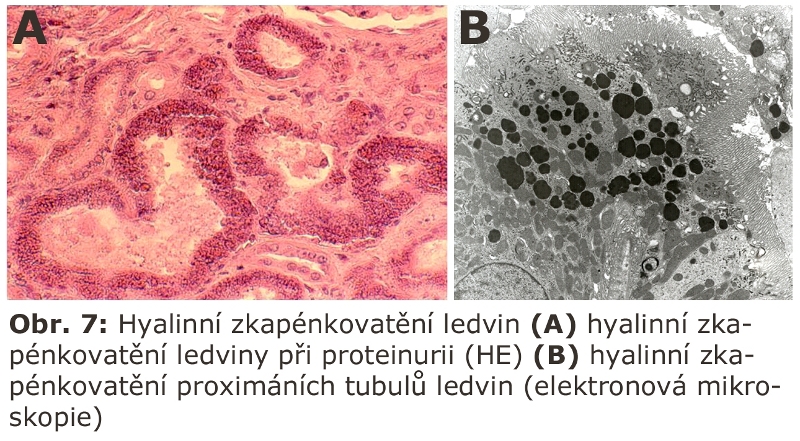
Lze zobecnit, že jakákoliv vysokomolekulární látka přítomná v nadbytku v primární moči se může mechanismem endocytosy (event. pinocytosy) dostat do lysosomálního systému v této části ledvin a hromadit se v něm.
Hyperlipoproteinemie (dyslipoproteinemie) mohou za určitých okolností vyvolat obraz generalisovaného střádání, postihující mononukleárně fagocytární systém, takže může být obraz, který připomíná do určité míry primární lysosomální střádací onemocnění. Typický je tento obraz u hyperlipoproteinemie typu I (Obr. 8).

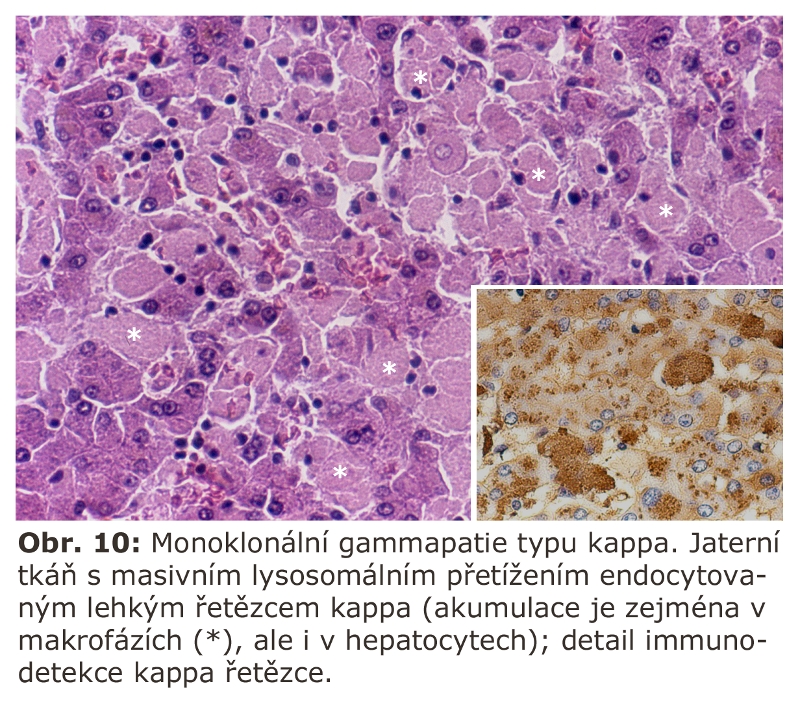
Některé monoklonální gammapatie z lehkých řetězců mohou vést ke generalisované endocytose příslušných řetězců, a to nejen makrofágy, ale i řadou dalších buněk. Je to nejspíše dáno tím, že abnormální lehký řetězec má doménu, která je ligandem pro endocytosu na buňkách nezávisle na jejich typu. Jako příklad uvádíme gammapatii z lehkých řetězců typu kappa (Obr. 9, 10).

Zde nebude věnována pozornost lokálním procesům, ve kterých jsou makrofágy aktivovány za účelem resorpce regresivně změněné, vesměs nekrotické tkáně. Detailně o většině z nich je pojednáno v samostatné kapitole Steatosy.
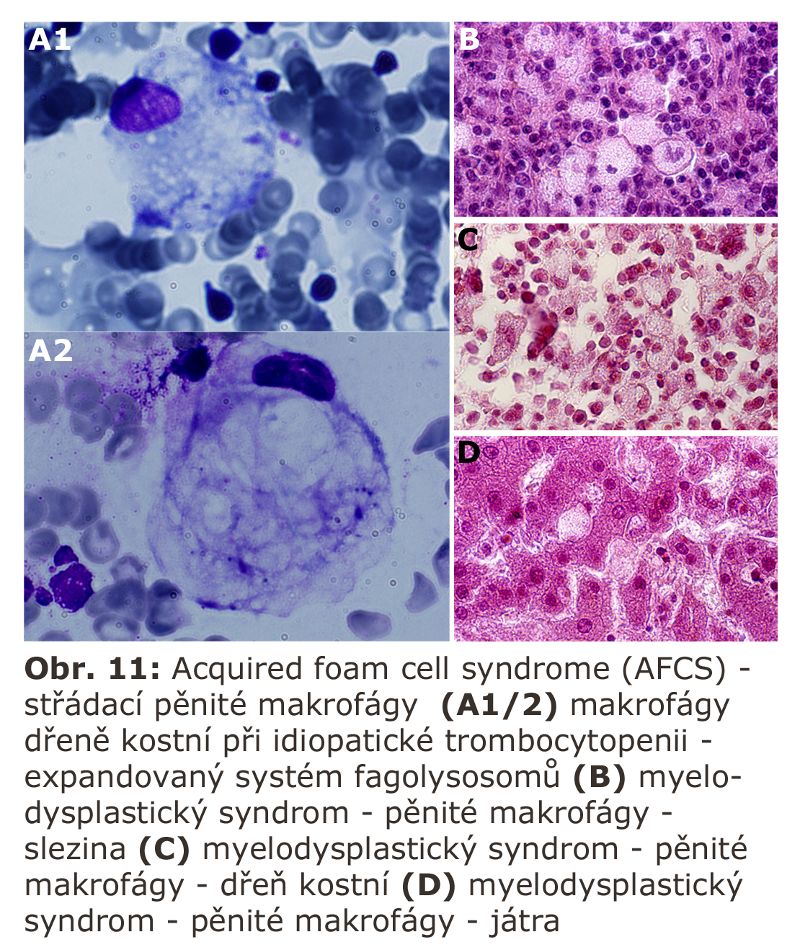
Zde přicházejí v úvahu hlavně celkové stavy s klinickou odezvou, vyznačující se vystupňovanou hemofagií, tedy fagocytosou krevních destiček, erytrocytů, případně leukocytů. Jde o trombocytopenii či hemofagickou lymfohistiocytosu, u kterých je jedním z projevů i splenomegalie (detailní specifikace těchto jednotek je v hematologických textech). Reaktivním buněčným typem jsou tkáňově rezidentní makrofágy - profesionální fagocyty, u kterých může dojít k rozvoji cytologických projevů neodlišitelných od lysosomálního střádání - tvorba pěnitých buněk a to v řadě orgánů (slezina, játra, dřeň kostní) (Obr. 11). Za tvorbu vakuol jsou zodpovědny hlavně fagolysosomy. Ostatní buněčné typy nejsou postiženy. Při detailním vyšetření je patrné různě vystupňované lysosomální střádání fosfolipidů a glykolipidů, případně cholesterolu. Při dlouhodobém přetížení ELS dochází velmi často i k deposici ceroidu.
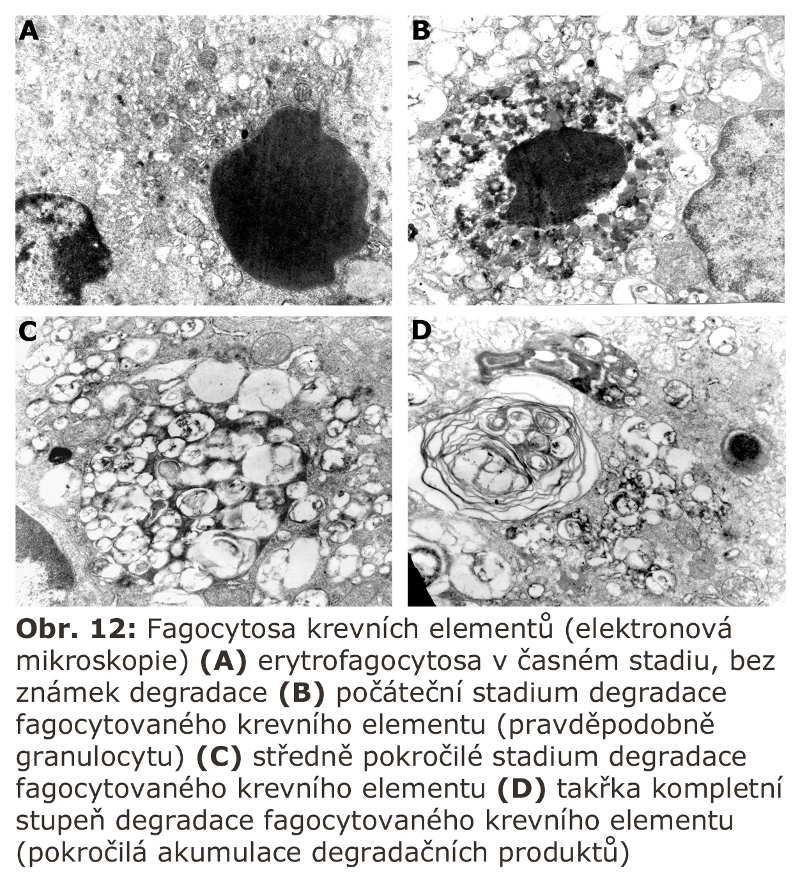
Elektronová mikroskopie ukáže fagocytované krevní elementy v různém stadiu degradace (Obr. 12).
V původní koncepci byly lysosomy považovány za "sebevražedné váčky", po jejichž disrupci dochází k nekontrolovanému uvolnění lysosomálních hydrolás do cytoplasmy s její následnou destrukcí. Existence tohoto mechanismu je stále předpokládána, i když některé fenomény byly vysvětleny jiným mechanismem. Například přítomnost extralyososomálních hydrolas v ischemicky poškozených tkáních byla zčásti vysvětlena větší fragilitou lysosomů a únikem hydrolytických enzymů v průběhu jejich isolace. Disrupce lysosomů je však stále pokládána za důležitý faktor v patogenesi onemocnění spojených s deposicí mikrokrystalů (microcrystalline diseases), zejména anorganických, např. SiO2 (u silikosy) nebo kalcium pyrofosfátu (vápenná dna), ale i organických (krystalu urátu u dny). Je otázkou zda dochází k disrupci přetížených lysosomů u lysosomálních střádacích onemocnění (viz níže).
Jde o velkou skupinu lysosomálních poruch, tzv. střádacích onemocnění (viz výše). První významnou podskupinou lysosomálních střádacích onemocnění jsou lysosomální enzymopatie, které se projevují progresivní expansí lysosomálního systému díky hromadění substrátu deficitního enzymu. Morfologicky (na úrovni světelné a elektronové mikroskopie) dochází k intralysosomální kumulaci těchto látek a následné expansi buněk a orgánů - z toho historický název střádací onemocnění.
Druhou podskupinou jsou geneticky podmíněné dysfunkce některého z neenzymových proteinů ELS. I v těchto situacích se však molekulární deficit projevuje na úrovni buňky, s určitými výjimkami podobně (konkrétně viz níže).
V současnosti je známo přes 50 molekulárně geneticky definovaných jednotek patřících do této skupiny. Není pochyb o tom, že se toto číslo bude v budoucnu zvyšovat. Omezeně probádanou je možnost existence lysosomálních poruch, neprojevujících se klasickými změnami typu "střádání". Jak bylo uvedeno úvodem, komplexita intracelulárních kontaktů a vztahů ELS a souvisejicích drah (např. autofagie) je enormní. Je proto otázkou zda nebude třeba komplexně přehodnotit pohled na patologii lysosomálního aparátu. Již nyní je jasné, že klasické enzymopatie vedoucí k fenotypu střádání z jasně definovaných katalytických příčin tvoří pouhou frakci celkového počtu všech uvedených jednotek. Je také jasné, že některá onemocnění řazená mezi lysosomální střádací onemocnění nejsou klasicky střádací onemocnění, ale spíše se jedná o onemocnění z lysosomální dysfunkce (typicky porucha clearence autofagosomů - Danonova choroba). V tomto smyslu je vhodné zmínit kupříkladu Smith-Lemli-Opitzův syndrom, u kterého byly popsány sekundární lysosomální změny velmi blízké Niemann-Pickově chorobě typ C. Pokud by měl být přijat tento zobecňující pohled na lysosomální patologii, nutně pak bude nutné hovořit i o stavech jako jsou např. Alzheimerova nebo Parkinsonova porucha jako o poruchách lysosomálních.
I ve skupině lysosomálních enzymopatií dochází k objevům nových deficitů, naprosto recentně byla popsána nová lidská patologická jednotka - cystická leukoencefalopatie na podkladě deficitu proteinu RNAasy t2 (gen RNASET2) štěpícího jednovláknové RNA (zřejmě lysosomálně). Jedná se o kongenitální defekt vývoje a funkce mozku, který je klinicky a radiologicky prakticky neodlišitelný od kongenitální cytomegalovirové infekce.Molekulární podstatou tohoto stavu je absence enzymatické aktivity jedné z lysosomálních RNAs (viz výše), lze očekávat, že se bude jednat o poruchu s lysosomálním střádáním nukleových kyselin (RNA) resp. střádáním ribonukloproteinů.
Je obtížné stávající lysosomální střádací onemocnění definovat obecně. Je jisté, že celá řada znaků je společná pro větší počet jednotlivých nosologických jednotek na úrovni buněčné, přes molekulární a biochemickou heterogenitu. Není účelem tohoto textu poskytnout vyčerpávající individuální popis molekulární podstaty (z úrovně DNA až po biochemii) každé jedné z těchto poruch. Naopak, cílem je představit určitý zobecňující pohled na buněčnou a orgánovou patologii lysosomálních geneticky podmíněných poruch (Schéma 8).
První velkou skupinu tvoří lysosomální střádací onemocnění, jejíchž společným jmenovatelem je deficitní katalytická funkce některého z lysosomálních enzymů. Jde celkem o 30 jednotek (29 isolovaných deficitů lysosomálních hydrolas a 1 deficit lysosomální transferasy).
Tuto skupinu je dále možné dělit na tyto podskupiny:
(i) deficity enzymů štěpících převážně lipidy - lipidosy (10 jednotek) (Schéma 9)
V případě deficitu lysosomální α-glukosidasy je zdroj substrátu normální proces autofagie (neexistuje přímý transmembránový transport glykogenu do ELS). K nadměrné zátěži lysosomálního aparátu může dojít abnormálním zvýšením makroautofagie. V kosterním svalu u Pompeho nemoci je, z ne zcela jasných důvodů, autofagie tak vystupňována, že dominuje nad střádáním glykogenu.
(v) deficit lysosomálních proteas (3 jednotky)U těchto deficitů dochází v lysosomech k akumulaci proteinů, které neodpovídají deficitní protease (viz níže)
deficit katepsinu A, fungujícího také jako protektivní protein pro komplex sialidasy a β-galaktosidasy. Biochemicky tedy jde o kombinovanou deficienci β-galaktosidasy a sialidasy (souhrnně je deficit katepsinu A nazýván galaktosialidosa).
Skupina katepsinů. Jde o skupinu čítající minimálně 13 proteas. Všeobecně se má za to, že katepsiny vykonávají proteolytickou funkci v ELS, některé z nich v oblasti časného endosomu, některé extracelulárně. Jejich proteolytická aktivita je zaměřena i na regulovanou proteolysu, pomocí které dochází k aktivaci celé řady proteinů (katalytických i nekatalytických). O jejich deficitech není mnoho známo.
O katepsinu A a D, resp. o stavu, ke kterému vede jejich mutace, byla již zmínka (viz výše).
Zvláštním případem je deficit katepsinu K, který je selektivně exprimován a secernován osteoklasty). Je zodpovědný za degradaci kostní matrix (kolagen typ I) a udržuje tak rovnováhu mezi tvorbou a degradací kostní tkáně. Důsledkem jeho mutace je tzv. pyknodysostosa. Degraduje velmi efektivně i elastin.
Deficit katepsinu C (dipeptidyl peptidasy I). Tento katepsin je exprimovaný zejména v makrofázích, polynukleárních leukocytech, ale i v dlaždicovém epitelu. Velmi pravděpodobně hraje zásadní roli v aktivaci dalších proteas v imunokompetentních buňkách. V mutovaném stavu je zodpovědný za Papillon-Lefevre syndrom charakterisovaný palmoplantární hyperkeratosou a těžkou periodontitidou, vedoucí k vypadávání zubů již v dětském věku. O klasickém lysosomálním střádání u této jedotky není nic známo.
U myší vedlo vyřazení katepsinu F k obrazu neuronální ceroid lipofuscinosy. Podobný fenotyp mělo u myší i kombinované vyřazení katepsinů B a L
(i) deficit formylglycin syntasy; formylglycin syntasa je enzym katalysující v endoplasmatickém retikulu vytvoření aktivního centra enzymů ze skupiny sulfatas (výsledkem jeho deficitu je tzv. polysulfatasový deficit, v němž se kombinuje deficit arylfulfatasy A s enzymatickými deficity ze skupiny mukopolysacharidos, viz výše - posttranslační modifikace ELS proteinů)
(ii) deficit enzymového komplexu (fosfotransferasa + fosfoglykosidasa) v terminální části Golgi aparátu, který katalysuje vytvoření M-6-P značky (mukolipidosa II). Postranslační modifikace formou M-6-P glykosylace je nezbytná pro koordinovaný a cílený transport drtivé většiny solubilních lysosomálních enzymů do lysosomů (viz výše). Důsledkem chybění M-6-P značky v glykosylačním vzorci lysosomálních hydrolas probíhá místo jejich fysiologického transportu ve vazbě na MPR z trans-Golgi do pozdních endosomů (viz výše) exocytosa do extracelulárního prostoru, kde jsou jejich aktivity mnohonásobně zvýšené a kde je lze také diagnosticky prokázat (např. v plasmě). Zpět do lysosomů se lysosomální hydrolasy bez M-6-P značky mohou dostat pouze endocytosou (pravděpodobně hlavně jako ligandy manosového receptoru). Tento mechanismus může zčásti kompensovat jejich intralysosomální deficit způsobený poruchou jejich intracelulárního cílení.
Pozn. pro lepší orientaci - mukolipidosa (ML) je historický termín naznačující heterogenitu střádání při použití klasických histochemických technik. Pro přehled shrnujeme, že původní ML I byla definována jako deficit sialidasy (viz výše), ML II je defekt posttranslační modifikace lysosomálních enzymů (defekt vytváření M-6-P značky), ML III jako alelická varianta MLII s pomalejším klinickým průběhem a ML IV je důsledek mutací v genu pro mukolipin (viz níže)
Schéma 10 podává přehled lysosomálních enzymopatií:

Druhou velkou skupinou dědičných lysosomálních poruch jsou onemocnění podmíněná mutací některého z lysosomálních (nebo funkčně s lysosomy souvisejících) proteinů bez enzymatické katalytické funkce (12 jednotek)
a) deficity membránových transportérů degradačních produktů přes lysosomální limitující membránu. (2 jednotky)
deficit transportéru pro sialovou kyselinu (výsledkem je akumulace kyseliny sialové v lysosomech)
deficit transportéru pro cystin (výsledkem je cystinosa charakterizovaná akumulací krystalků cystinu v lysosomech)
b) deficit transportu lipidů přes lysosomální membránu (2 jednotky)
m. Niemann-Pick typ C (porucha transportu lipidů spojená s lysosomálních akumulací glykolipidů, sfingomyelinu a cholesterolu). Stejný klinický i buněčně patologický fenotyp je způsoben mutacemi v jednom ze dvou NPC genů (NPC1 a NPC2). Mutace v genu NPC1 představují dominantní komplementační skupinu m. Niemann-Pick typ C (přítomny u 95% všech pacientů s Niemann-Pickovou chorobou typ C), mutace v NPC2 genu jsou minoritní (5 % pacientů) komplementační skupinou. NPC1 a NPC2 proteiny funkčně kooperují a zprostředkovávají transport neesterifikovaného cholesterolu přes lysosomální membránu zpět do cytosolu. NPC1 je integrální membránový protein lysosomální limitující membrány, NPC2 (podstatně menší protein než NPC1) je naopak solubilní luminální lysosomální protein. Oba proteiny mají schopnost vázat cholesterol a koordinovaně si jej v průběhu translokace přes lysosomální limitující membránu předávat.
c) deficit mukolipinu (mukolipidosa IV, ML IV)
Funkce mukolipinu není doposud zcela jasná, navržena byla funkce tohoto proteinu v intralysosomální homeostáze kalcia; důsledkem deficitu mukolipinu je heterogenní střádání lipidních a nelipidních látek v lysosomech, postiženy jsou i parietální bb. žaludeční sliznice
d) deficit proteinu LAMP2 (lysosomal associated membrane protein 2) - Danonova nemoc
Jde o lysosomální dysfunkci týkající se autofagie (zejména makroautofagie), která se projevuje defektním intracelulárním zpracováním autofagosomů v řadě buněk, zejména v kardiomyocytech a myocytech kosterních svalů. LAMP2 zprostředkovává komunikaci a fúzi autofagosomů (ale i fagosomů) s lysosomy. Sestřihová varianta "A" LAMP2 proteinu (LAMP2A) je receptorem tzv. "chaperony" zprostředkované autofagie (viz výše). LAMP2 má dále schopnost interagovat s mikrotubuly a jejich asociovanými proteiny. Lze předpokládat, že tímto způsobem zajištuje správnou intracelulární distribuci lysosomů a autofagosomů.
Poznámka. Nutno zmínit, že existují i stavy s patologicky rozvinutou autofagocytosou, které nejsou doposud přesně definovány na buněčně patologické úrovni. Mezi postižené patří zejména tkáň příčně pruhovaného svalu. Výjimkou mezi těmito stavy je deficit GNE (UDP-N-acetylglucosamin 2-epimerasa/N-acetylmanosamin kinasa). Vztah tohoto proteinu k autofagii však není doposud spolehlivě definován.
c) deficit LIMP2 proteinu (lysosomal integral membrane protein 2)
Jde o novou, nedávno popsanou, nozologickou jednotku. V klinickém obraze tohoto stavu dominuje myoklonická epilepsii, kombinovaná v některých případech se selháním ledvin, souhrnně nazýváno AMRF syndrom (action myoclonus renal failure syndrome). Biochemicky je deficit LIMP2 spojen s deficitem β-glukocerebrosidasy, jelikož tato je do lysosomů transportována právě v asociaci s LIMP2 proteinem (viz výše). Tato jednotka je v současnosti předmětem intensivního výzkumu
d) skupina neuronálních ceroid lipofuscinos - NCL (7 definovaných jednotek NCL3, 5, 6, 7, 8, deficit CLC7 a Ostm1, viz níže). Nejsou zde započítány typy NCL způsobené enzymopatiemi, tj. NCL1, NCL2, NCL10)
Tato skupina je názorným příkladem nedostatečnosti našich současných znalostí o procesu formálně, strukturálně a geneticky dobře definovaném (s výjimkou adultní formy NCL4 historicky zvané m. Kufs). U všech níže uvedených jednotek jsou známy geny, kodující proteiny (bez enzymatické aktivity), které v mutantní formě způsobí fatální neurologické poruchy, jejichž společným projevem je neurolysosomální střádání, jehož mechanismus není stále adekvátně vysvětlen. Funkce těchto proteinů za normální situace není známa. Podobně není znám mechanismus, kterým se stává lysosomální systém defektní při jejich dysfunkci způsobené vrozenou mutací.
NCL3 protein je integrální protein lysosomální membrány, velmi pravděpodobně polyfunkční, s výraznou expresí v synaptosomech. Jeho mutace vede k NCL převážně juvenilního typu (m.Batten-Spielmeyer-Vogt)
NCL5 protein je lokalizován v lysosomech a je považován za solubilní; jeho mutace vede k pozdně infantilní formě NCL
NCL6 protein je lokalisován v ER. Jeho mutace vede jako u NCL5 k pozdně infantilní formě NCL
NCL7 je lysosomální membránový protein. Jeho mutace vede k pozdně infantilní formě NCL
NCL8 protein je lokalisován v ER i v Golgiho aparátu. Jeho mutace vede k časné formě NCL s epilepsií, mentální retardací a poruchou motoriky; je též zvaná "severská epilepsie"
(NCL9 čeká na upřesnění genetické poruchy - biochemicky jde o deficit regulátoru dihydroceramid syntasy)
Před několika lety se vyčlenila zvláštní skupina neuronálních ceroid lipofuscinos, kombinovaných s kostními změnami ve smyslu osteopetrosy. Jde o mutace ve skupině chloridových kanálů (CLC chloride channels), účastnících se na acidifikaci lysosomů a exprimovaných v osteoklastech, kde zajišťuje acidifikaci míst degradace kostní matrix. Mutace v proteinu CLC -7 (nebo v proteinu Ostm1, který je ve funkčním komplexu s CLC-7) byla prokázána jako zodpovědná za lidská onemocnění.
U většiny jednotek NCL (s výjimkou NCL1 a NCL10) se v lysosomech akumuluje hyper-hydrofobní protein fysiologicky fungující v rámci vnitřní mitochondriální membrány. Jedná se o podjednotku c komplexu V (ATP syntasa) kaskády oxidativní fosforylace (OXFOS) (Schéma 11). Zvláštností tohoto proteinu je extrémní hydrofobicita a rozpustnost v organických rozpouštědlech. Stupněm své hydrofobicity převyšuje nepolární lipidy.
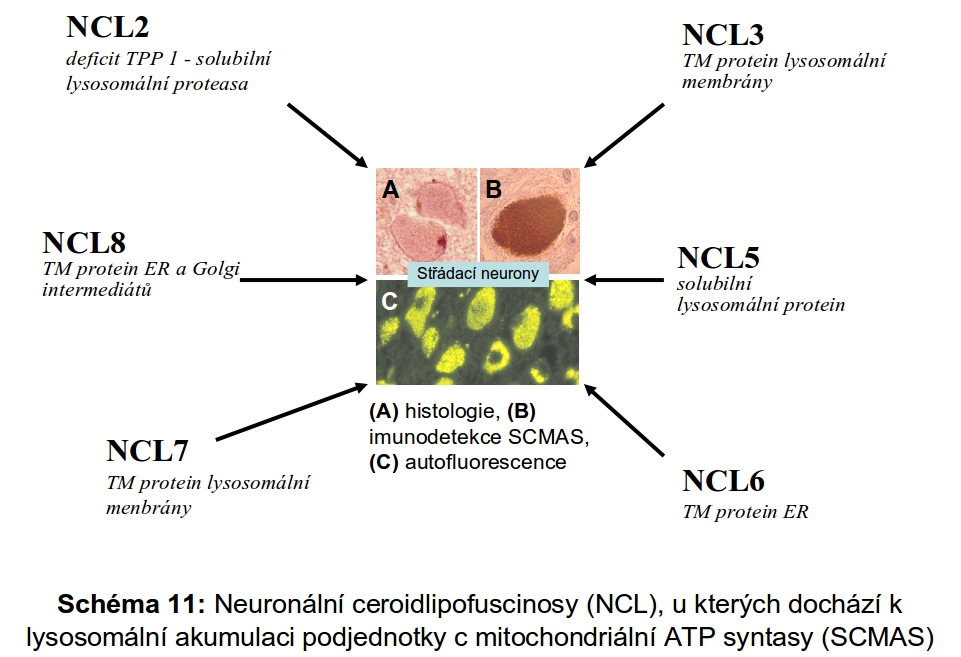
Přehled této skupiny lysosomálních střádacích poruch na podkladě neenzymatického proteinového deficitu je podán ve Schématu 12

1. celou původní molekulu (např. lipid), jestliže je deficitní enzym, který je prvý v degradační řadě enzymů a jehož deficience blokuje následnou funkci dalších enzymů v metabolické degradační řadě. Případný deficit takových (následně fungujících) enzymů se pak může projevit jen rudimentárně. Ne zcela vyjasněný je vznik lysoderivátů (glukosylceramidu u Gaucherovy nemoci či galaktosylceramidu u Krabbeho nemoci), které jsou výrazně cytotoxické. Mnoha autory jsou tyto formy považovány za produkt abnormální syntesy.
2. mezistupeň v souvislé degradační řadě (typické pro degradované glykosfingolipidy s delším cukerným řetězcem). Iniciální substrát je fysiologicky parciálně degradován až po kritickou, avšak v kontextu enzymatického deficitu neštěpitelnou vazbu. K největší modifikaci primárního substrátu dochází u deficitů aktivity glykosidas a sulfatas, jejichž substráty jsou dlouhé řetězce glykoproteinů a glykosaminoglykanů. Zde je výchozí vysokomolekulární substrát sukcesivně zpracováván řadou enzymů (exo- i endoglykosidas resp. peptidas). Důsledkem je pak akumulace různě velkých fragmentů obsahujících kritickou vazbu.
3. diskrepance. V některých případech se v lysosomech hromadí látky neodpovídající deficitnímu enzymu. Jde typicky o neurolysosomy ve skupině mukopolysacharidos typ I-III, jimž je společné defektní odbourávání heparan sulfátu. V neuronech se hromadí směs gangliosidů a některých vyšších ceramidhexosidů spolu s autofluorescentním lipopigmentem (viz níže). Vysvětlení této odlišné biochemické reakce ve srovnání s epitelovými a mesenchymálními buňkami chybí.
Biologicky velmi zajímavou situaci představuje deficience α-N-Acetyl-galaktosaminidasy (koncový cukr u krevní skupiny A) u tzv. Schindlerovy nemoci. Zde může být dokonce dominantním neuropatologickým příznakem u některých pacientů rozvoj obrazu extrémní neuroaxonální dystrofie (defekt axonálního transportu neznámého původu), aniž by byly patrny známky lysosomálního střádání kritických glykokonjugátů.
Stále nevyjasněná je situace u Gaucherovy nemoci, u které je lysosomální střádání vyjádřeno pouze v jediném buněčném typu - v makrofágu. V žádné jiné buňce nebylo v lysosomech střádání pozorováno. Ani v neuronech u těžké neuronopatické formy (GD II), u které neurony degenerují bez známek lysosomálního střádání.
Zcela nejasný je vztah mezi enzymovým deficitem u NCL1 (deficitní je palmitoyl protein thioesterasa, která odštěpuje mastné kyseliny thioestericky vázané na -SH skupiny jiných proteinů) a akumulací saposinu A a C (proteiny) v lysosomech. Při deficitu katepsinu D (NCL 10) jsou v lysosomech prokazatelné opět některé saposiny. V případě NCL2 (deficit exopeptidasy tripeptidyl peptidasy I) se kumuluje v lysosomech velké množství podjednotky c mitochondriální ATP syntasy (součást komplex V OXFOS). Podobně v dalších jednotkách NCL je diskrepance mezi charakterem deficitního proteinu a akumulovaným autofluorescentním materiálem.
Vedle hlavních nedegradovaných substrátů se mohou hromadit některé látky i sekundárně, např. cholesterol u řady lipidos, glykosfingolipidy u deficitu kyselé sfingomyelinasy (m. Niemann-Pick typ A a B) či laktosyceramid u řady lysosomálních poruch.
V omezené míře je vysvětlitelný vztah mezi funkcí mutovaných proteinů a lysosomálním střádáním ve druhé skupině lysosomálních poruch, u kterých není deficitní definovaná enzymová aktivita. Výjimku zde tvoří pouze defekty transportérů (pro sialovou kyselinu a cystin) a částečně též deficity NPC1 a NPC2 proteinů u Niemann-Pickovy choroby typ C. Zde je alespoň částečně jasný vztah k akumulaci resp. intracelulární poruše distribuce neesterifikovaného cholesterolu (viz výše - funkce NPC1 a NPC2 proteinů). Nezodpovězenou otázkou však zůstává paralelní lysosomální akumulace glykosfingolipidů (GM2, GM3), která v určitých buněčných typech (neurony) dominuje nad střádáním cholesterolu. Přitom je zajímavé, že enzymy nutné k degradaci těchto lipidů nemají biochemicky prokazatelně sníženou aktivitu.
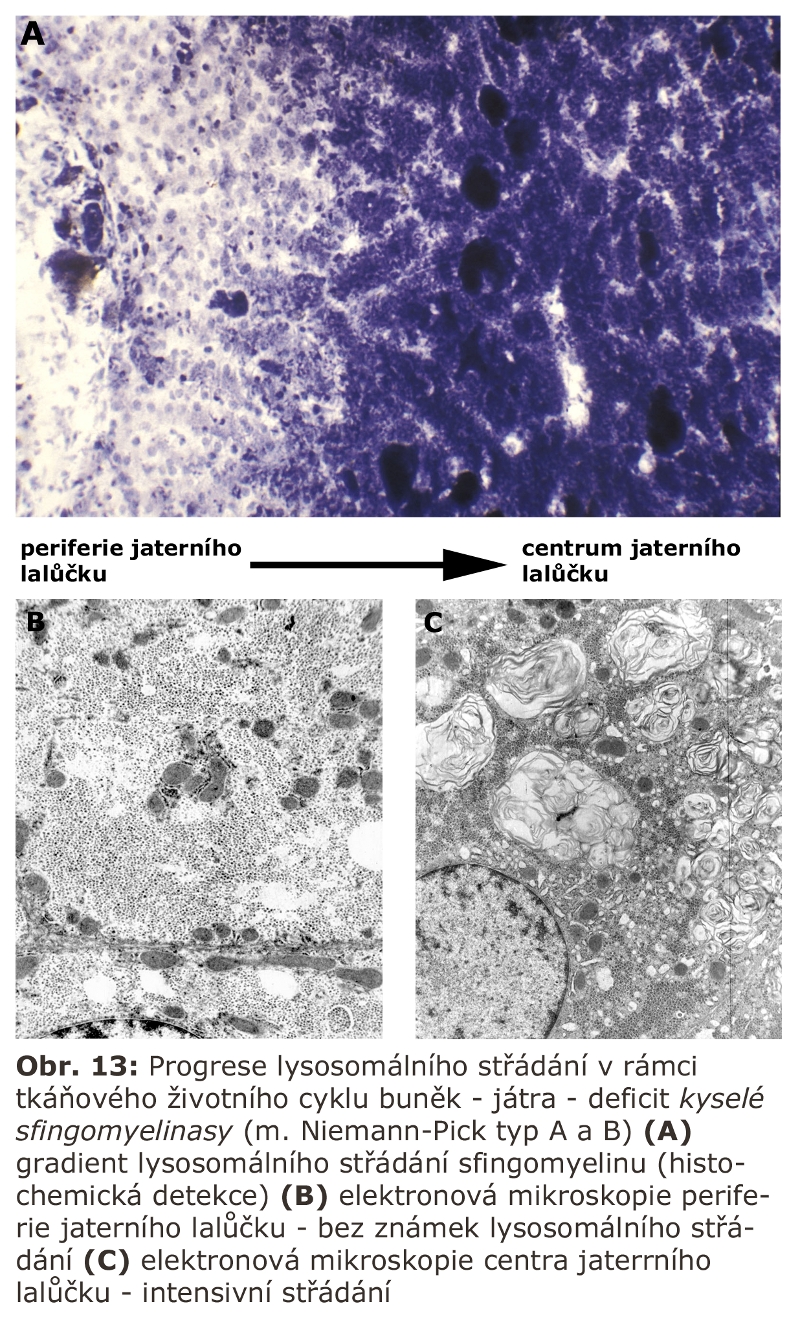
Je nutné si uvědomit, že za popisným fenomenem lysosomálního střádání stojí narušení normální biologie buňky. Normální buňka se tak stává buňkou postiženou lysosomálním střádáním, která je v mnoha rysech dysfunkční. Primárním důvodem je zřejmě dysfunkce ELS, jehož běžné funkce jsou komplexně narušeny, což vyvolává kaskádu následných dějů, které jsme v současnosti schopni více či méně logicky propojit s primárním genetickým deficitem ELS. Zkušenost ukazuje, že všechny stavy spojené s lysosomální expansí jsou v zásadě nekompatibilní s vitalitou buňky. Nadměrná a nekompensovaná distense lysosomálního systému interferuje doposud nejasným způsobem s vitalitou buňky a s celou řadou fyziologických buněčných funkcí. Za závažný a možný důsledek lysosomálního střádání se považuje i disrupce lysosomální limitující membrány spojená se ztrátou integrity. Distense lysosomálního systému není omezena na nedegradované enzymové substráty, ale i na zadržené enzymové produkty, a v mnoha závažných případech i na retenci intaktních proteinů, peptidů či dalších molekul, bez známek jejich prokazatelné degradace. Do projevů dysfunkce ELS patří dnes již historický fenomén indukce tvorby tzv. ceroidu ve střádacích lysosomech, což je doposud nevyjasněný fenomén (viz níže - Residuální lysosomální tělíska)
Jako extrémní příklad inkompatibility je buněčná smrt na podkladě lysosomálního střádání (např. časný zánik oligodendroglie u Krabbeho nemoci - viz Schéma 13). To vede k nestabilitě myelinu a jeho odbourání makrofágy. Podobně je vysoká citlivost neuronů na střádací proces ve skupině neuronálních ceroid lipofuscinos či infantilní formy (typ II) m. Gaucher. Vedle efektů jasně souvisejících s lysosománím střádáním lze pozorovat i patologické změny zdánlivě s ELS funkcí nesouvisející. Typicky se jedná třeba o poruchu axonálního transportu formou neuroaxonální dystrofie u celé řady lysosomálních střádacích onemocnění. V případě neuroaxonální dystrofie si lze velmi dobře představit, že důsledkem této změny je například porucha retrográdního transportu neuronálních trofických faktorů což může zásadně interferovat s vitalitou neuronu. Co spojuje lysosomální střádání s dysregulací axonálního transportu je nejasné. Podobné je to i s důvody pro aktivaci mikrooglie v CNS v rámci probíhajícího lysosomálního střádání.
O narušené biologii buňky svědčí i zdánlivě progresivní změny (např. ektopická dendritogenese v oblasti iniciálního axonu), jak to bylo prokázáno v neuronech akumulujících GM2 gangliosid buď primárně (deficit β-hexosaminidasy) nebo sekundárně (m. Niemann-Pick typ C).
Nutno zmínit i, dnes již historický, fenomén indukce tvorby tzv. ceroidu, což je doposud nevyjasněný fenomén (viz oddíl Reziduální lysosomální tělíska).
Podstatou lysosomálního střádání je přeplnění lysosomálního aparátu nerozštěpeným substrátem či netransportovatelným produktem. U části lysosomálních střádacích onemocnění (skupina NCL) jsou v ELS, z důvodů v současnosti nejasných nebo jasných pouze částečně, akumulovány hydrofobní proteiny (typicky podjednotka C mitochondriální ATP syntasy, viz Schéma 11). Je jisté, že strukturální expanse lysosomálního aparátu vede k jeho dysfunkci, která je v různé míře nekompatibilní s fysiologií postižené buňky. Distense lysosomálního systému není tedy omezena na nedegradované enzymové substráty, ale i na zadržené enzymové produkty a v mnoha závažných případech i na retenci intaktních proteinů, peptidů či dalších molekul, bez známek jejich prokazatelné degradace. Celkově tedy dochází v rámci buňky k propagaci patologie způsobené deficitem individuálního genu a jeho proteinového produktu.
Závažnou skutečností, zejména ve skupině lipidos nebo mukopolysacharidos jsou paralelní akumulace jiných (typicky glykosfingolipidy a cholesterol) než primárně nedegradovaných substrátů. Zásadní je, že v průběhu střádání se mění lipidní kompozice nejen membrán ELS, ale i membrán jiných kompartmentů (Golgiho aparátu nebo cytoplasmatické membrány). Změnou kompozice membrán mimo ELS dochází k další propagaci systémového dopadu lysosomálního střádání.
Přestože je proces lysosomálního střádání u velké části uvedených jednotek vyvolán deficitem v degradaci a důsledkem je akumulace, není doposud jasné, zda postiženým buňkám více vadí ona excesivní akumulace substrátu nebo spíše vnitřní hladovění z důvodu dysregulace recyklace. Co je v tomto případě příčinou a co kompenzatorním důsledkem není jasné. V určitých situacích je paradoxně závažnější dysregulovaná recyklace.
Jak bylo řečeno úvodem této části, střádací lysosom není ekvivalentní k normálnímu lysosomu, stejně tak střádací buňka není buňkou normální. V současné době máme vcelku jasnou představu jakých intracelulárních drah a procesů se lysosomální střádání může v negativním smyslu dotknout (uvádíme stručný výklad, pro detailnější informace viz doporučená literatura).
Z uvedeného výčtu, byť vyznívá přesvědčivě, je možné říci, že buněčná patofyziologie následků lysosomálního střádání (dysfunkce) není z velké části vysvětlena, což je, znovu, v kontrastu s přesnou definicí (až na diagnostickou molekulárně genetickou úroveň) těchto chorob.
Vzhled buněk s nadměrně expandovaným lysosomálním systémem je dán nejen počtem a velikostí expandovaných lysosomů, ale i charakterem jejich obsahu.
S určitým omezením lze vnést dynamický pohled na rozvoj struktury ELS expandujícího střádáním. Doposud nejasné zůstávají mechanismy zásadním způsobem ovlivňující cytologický vzhled střádajících buněk. Jedná se o následujícíí aspekty (Obr. 14): (i) regulace velikosti individuální lysosomální vakuoly, kterou lze pokládat za nezávislou (samostatnou) strukturně biologickou podjednotku ELS, (ii) regulace počtu lysosomálních střádacích vakuol resp. jejich fúzí, které jsou velmi často morfologicky patrné, zvláště když dosahují velkých rozměrů. Nápadná je rozdílnost velikosti střádacích lysosomů, naznačující možnost fúze primárních vakuol nebo jejich nekontrolované zvětšování.
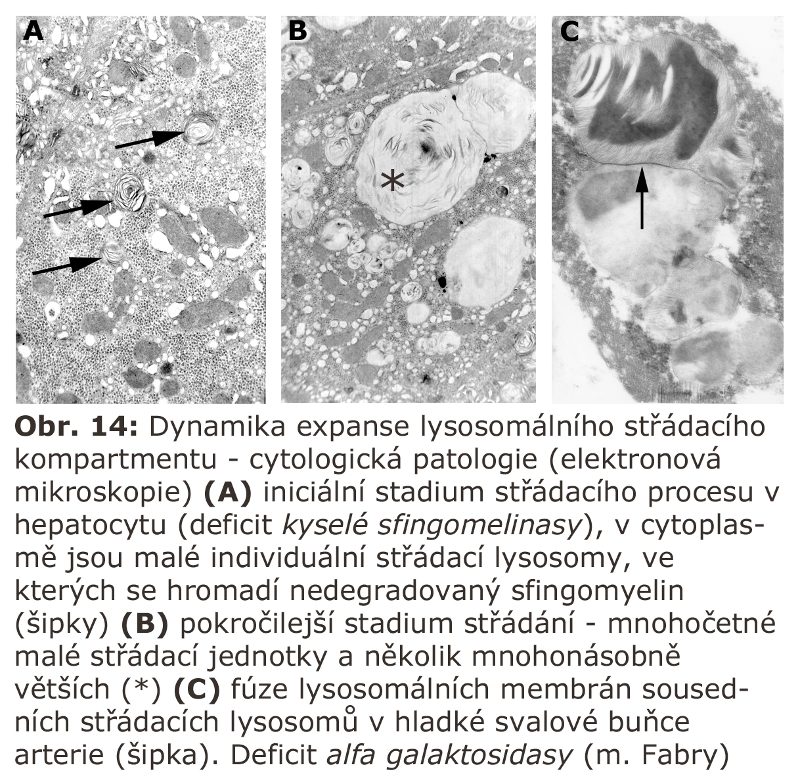
V některých případech dochází k vytváření velmi objemných variant co do velikosti, takže dominují, zvláště pokud není střádaný obsah odstraněn zpracováním jako proteinové sferoidní inkluse. Typicky je tomu v neuronech některých oblastí mozku u neuronálních ceroidlipofuscinos (Obr. 15).
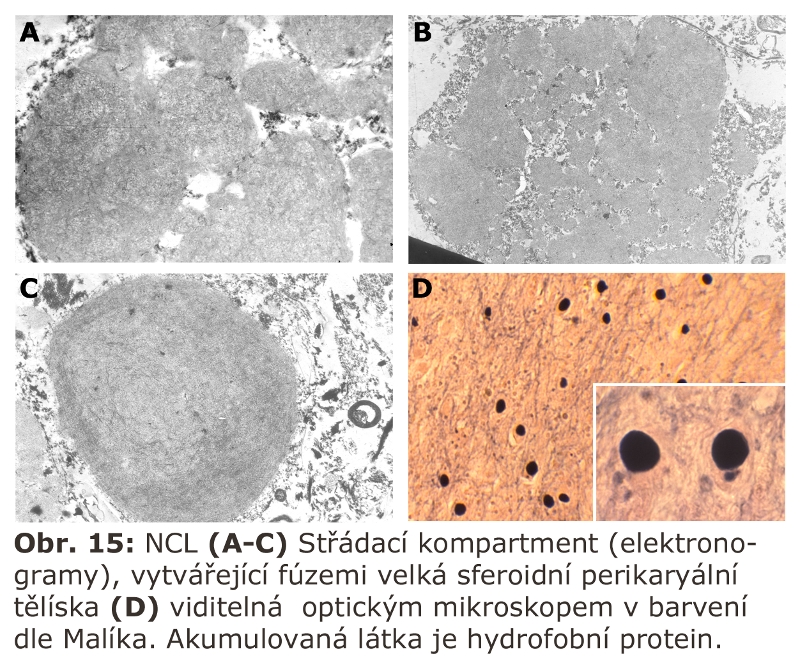
To kontrastuje se stavy, kdy i při pokročilém stupni střádání je zachována individualita střádacích lysosomů (Obr. 16)
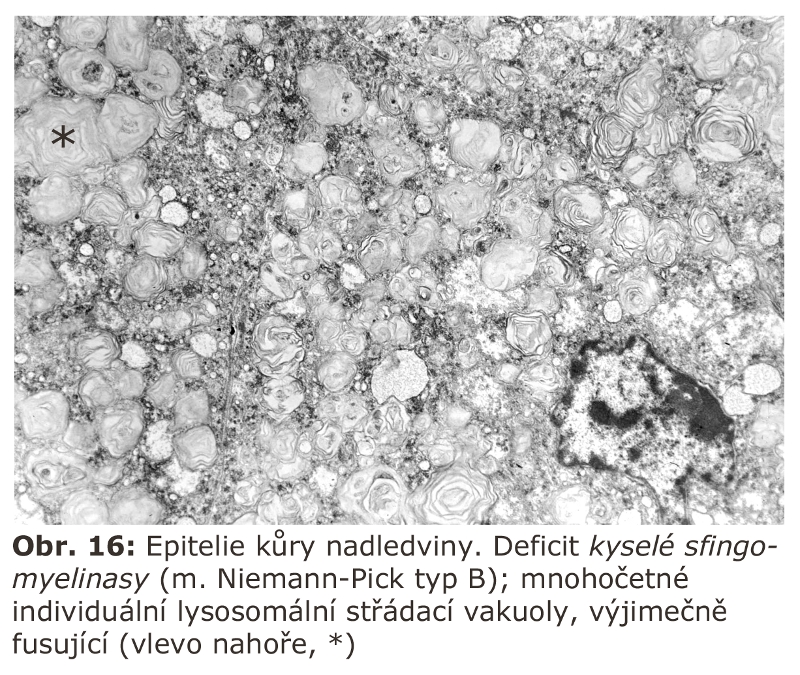
Souhrnně řečeno, morfologický vzhled střádací buňky je dán počtem a velikostí střádacích lysosomů.
Je-li střádaný biologický materiál solubilní v organických rozpustidlech (lipid) nebo solubilní ve vodném fixativu (glykopeptidy, oligosaccharidy), resultuje mikrovakuolisace až pěnovitá přeměna (voštinovité, pěnité buňky). Pokud se podaří obsah udržet in situ, je ve formě "kapének". Je-li produkt nesolubilní (lipopigment, glykogen, bakterie) mají buňky vzhled převážně granulovaný (jemná granula, větší hrudky, a pod.). Jiný vzhled může být daný tvarem bakterie (např. mykobakteria nebo Corynebakterium aequi u Whippleovy nemoci) nebo obecně tvarem fagocytovaného materiálu.
Existují zvláštní varianty cytologického vzhledu, dané protáhlým až štěrbinovitým tvarem lysosomů, které nezaplňují zcela cytoplasmu buňky. Takto morfologicky modifikované lysosomy pak dávají cytoplasmě zvláštní žíhaný vzhled, připomínající jemně pomačkaný papír. Tento fenomén je příznačný pro střádání cerebrosidů v makrofázích (Gaucherovy buňky) u deficitu glukocerebrosidasy (Gaucherova nemoc). Pokud jsou lysosomy volně dispergovány v cytoplasmě, jejíž organelové systémy jsou hyperplastické, může být celkový vzhled cytoplasmy solidní. To je typické pro střádací buňky mikroglie u Krabbeho nemoci - tzv. Krabbeho buňky připomínají epiteliodní histiocyty.
Srovnání hlavních cytologických projevů lysosomálního střádání je podán na Obrázku 17
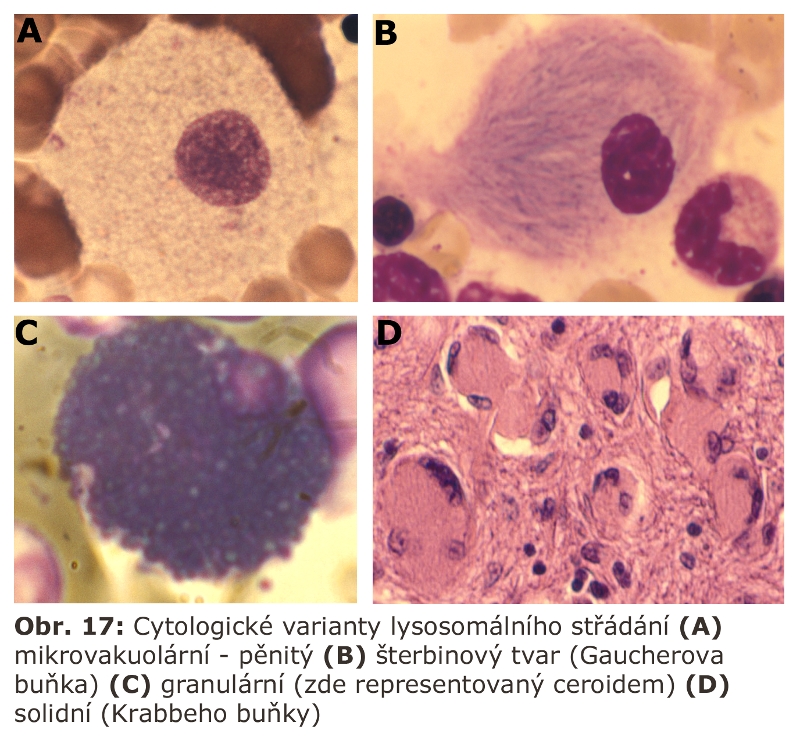
Při analyse střádáním postižených tkání je nutno počítat s paralelní deposicí primárně střádané látky (viz kapitola Lysosomální residuální tělíska)
Dlouhodobé trvání lysosomálního střádání může vést k výrazné kumulaci změn na buněčné úrovni, které se následně odrazí v rozvoji orgánových poruch, patrných již při standardním klinickém vyšetření.
Myokard. Manifestuje-li se lysosomální střádání v kardiomyocytech, je pravidelným důsledkem hypertrofická kardiomyopatie. Klasickým příkladem je deficit α-galaktosidasy (m. Fabry). Jako vyvolávající moment lze předpokládat sníženou kontraktilitu lysosomálním střádáním postiženého kardiomyocytu. Hypertrofii lze vysvětlit jako sekundární kompensační proces. Spolupostiženy bývají i kardiocyty převodního systému. Pokud jsou střádáním postiženy i koronární artérie rozvíjí se ischemické změny. U Fabryho nemoci je časté postižení chlopňového aparátu. Obrázek 18 shrnuje uvedené změny u m Fabry.
Lysosomální střádání v mesenchymálních elementech chlopně postihuje často srdce ve skupině onemocnění typu mukopolysacharidos. Vede k fibrose, deformaci a dysfunkci příslušné chlopně.
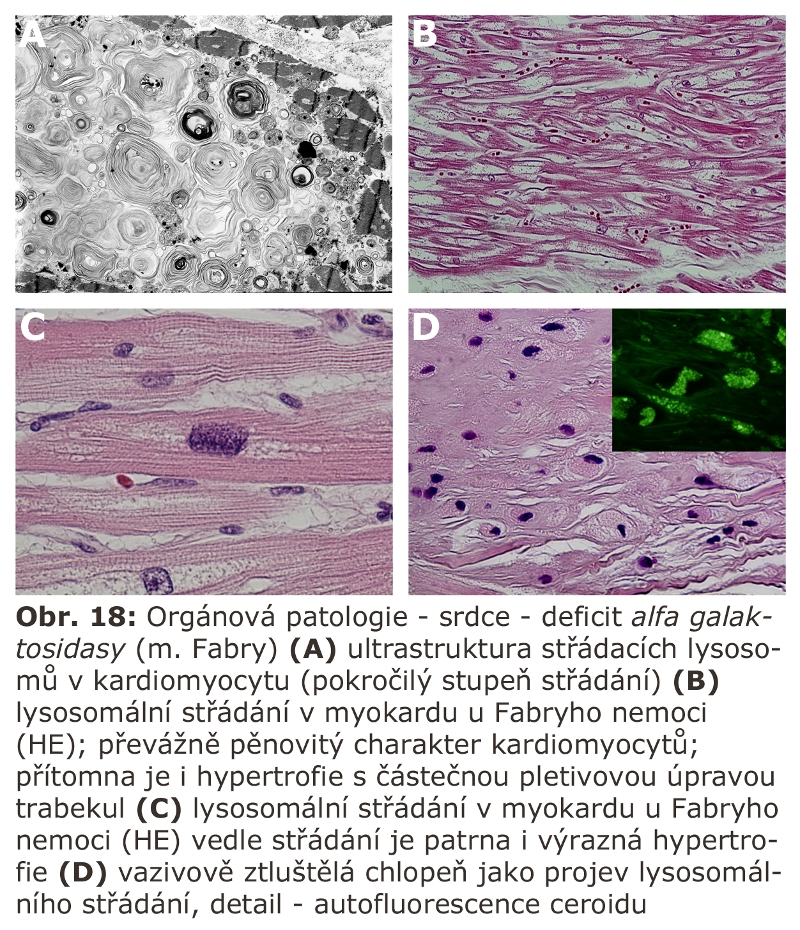
Ledviny. Postihne-li střádání výrazněji buňky renálních glomerulů, může dojít k poruše obratu komponent basální membrány kapilár a mezibuněčné hmoty mesangia, a následně k rozvoji proteinurie. Ke střádání inklinují zejména podocyty (Obr. 19 A, B). V dalším průběhu dochází k postupné hyalinní obliteraci glomerulu s následným selháním ledvin (Obr. 19 C). Běžné jsou tyto změny u Fabryho nemoci (deficit α-galaktosidasy). U Fabryho nemoci byl prokázán i úbytek uromodulinu a jeho abnormální intracelulární procesování (důsledek střádání v ascendentní části Henleho kličky, kde je tento protein produkován). Tubulární střádání je přítomno u sulfatidosy. Postižení tubulů se projevuje abnormálním složením lipidů v močovém sedimentu, čehož je využíváno pro diagnostiku jak u Fabryho nemoci(Obr 19D.), tak u sulfatidosy (Obr. 20).
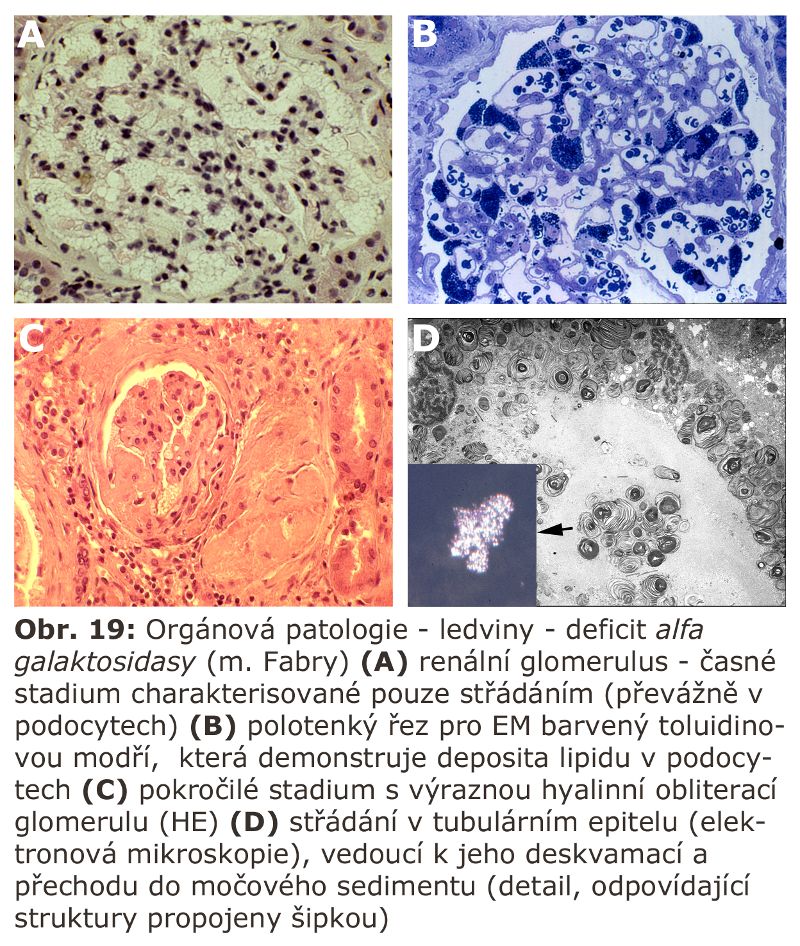
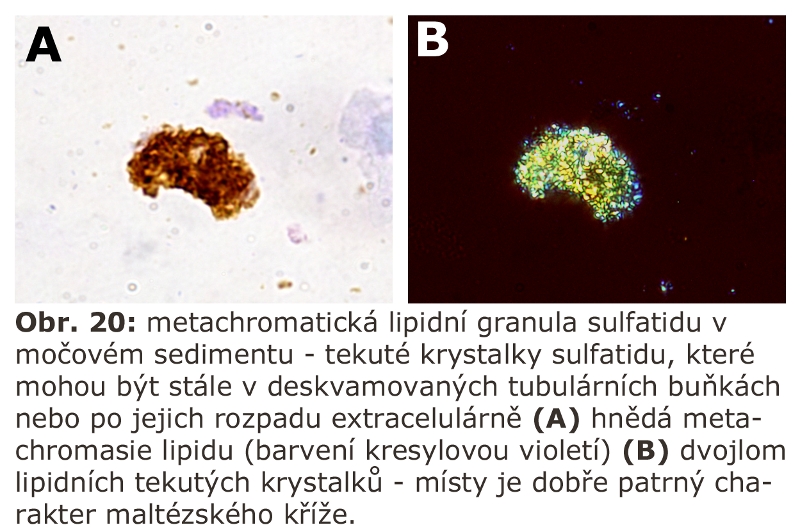
V případě postižení mesenchymu (fibrocyty, chondrocyty, osteocyty) je narušen obrat komponent extracelulární hmoty a může dojít ke zmnožení vaziva (pokud jsou postiženy chlopně srdeční, dojde k srdeční vadě) a k poruše harmonického vytváření tkání tvořených mesenchymem. Výsledkem je obraz dysmorfie či dysostosa (typicky u skupiny mukopolysacharidos).
Játra. U deficitu kyselé sfingomyelinasy a kyselé lipasy dochází k masivnímu střádání v hepatocytech, případně i v Kupfferových buňkách (Obr. 21). U mírně vyjádřených forem střádání nebo obecně v počátečních fázích je střádání vyznačeno v peribiliární oblasti hepatocytů, odkud se časem rozšiřuje na celou cytoplasmu. Může tak dojít k progresivnímu zániku hepatocytů s rozvojem fibrosy, event. cirhosy. Vzácně dochází k rychle progredujícímu jaternímu selhání, klasicky u infantilní formy Niemann-Pickovy choroby typ C. Střádání u Gaucherovy nemoci je omezené na Kupfferovy buňky (Obr. 22).
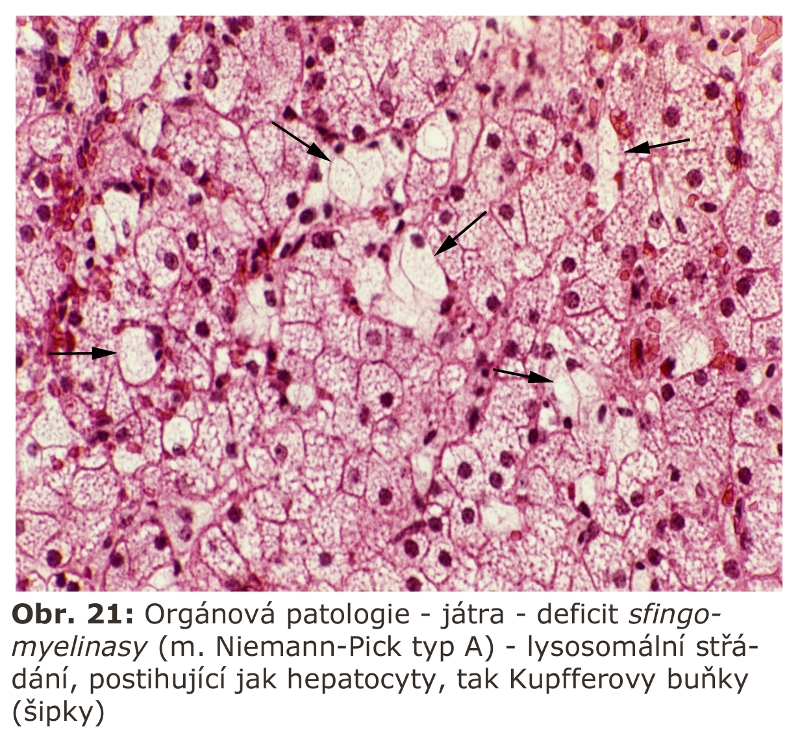
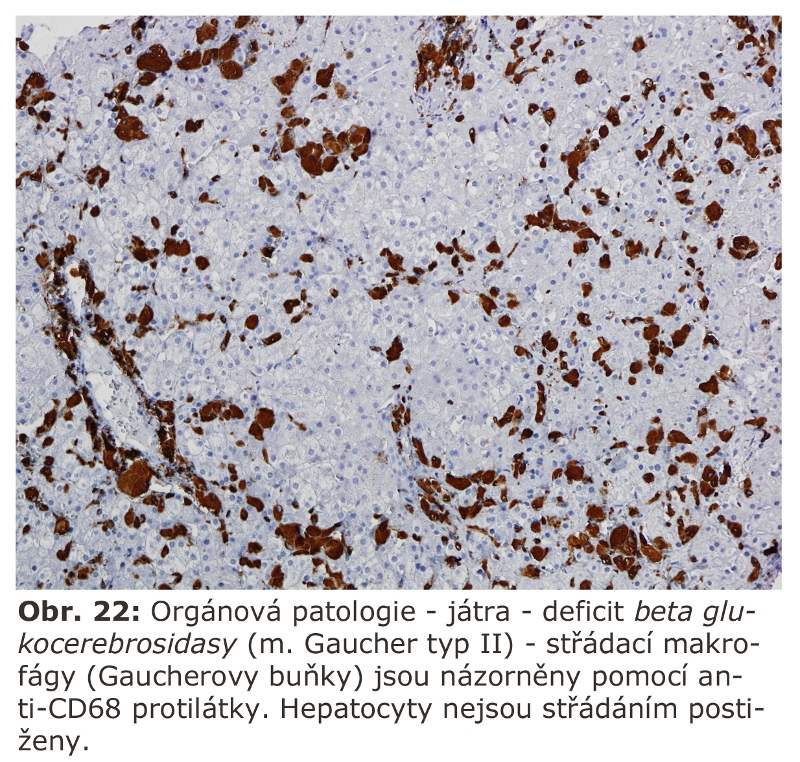
Mozek. Neurolysosomální střádání vede v počátečním stadiu k přeplnění perikarya neuronu s jeho "balonováním" (Obr. 23). V řadě případů dochází k manifestaci střádání i v iniciální části axonů (axon hillock) (Obr. 24 A, B). V případě Purkyňových buněk lze často pozorovat střádání i v oblasti dendritů, a to v různém poměru ke střádání v perikaryu (Obr. 24 D, C). Následuje postupný zánik neuronů (Obr. 25 B, C, D) a rozvoj astrogliosy (Obr. 25 E, F). Degenerující neurony jsou odstraněny mikrogliálními fagocyty za tvorby neuronofagických uzlíků. Na CT a MRI je hlavním nálezem postupná atrofie mozku (Obr. 25 A), případně i mozečku. U některých neurolysosomálních poruch dochází před rozvojem regresivní fáze neuronálního postižení k ektopické dendritogenesi v iniciálních oblastech axonů. Zřejmě se jedná o projev stimulace střádacího neuronu. Předpokládá se, že za stimulační efekt je zodpovědný primárně (u GM2 gangliosidosy) nebo sekundárně (např. u Niemann-Pickovy choroby typ C) akumulovaný GM2 gangliosid . K velmi výrazné sekundární změně může dojít i porušením axonálního toku a tvorbě tzv. axonálních sferoidů (neuroaxonální dystrofie, viz výše). Podklad rozvoje neuroaxonální dystrofie nalézané u celé řady lysosomálních střádacích onemocnění (např. Niemann-Pickova choroba typ C nebo α-manosidosa, ale i jiné) je naprosto nejasný, přestože se jedná o významnou a dlouhou dobu známou neuropatologickou změnu jednoznačně související s porušeným axonálním transportem.
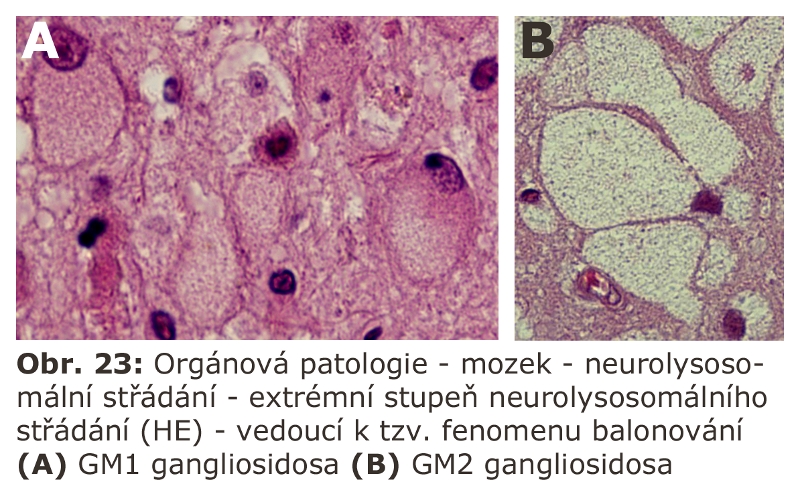

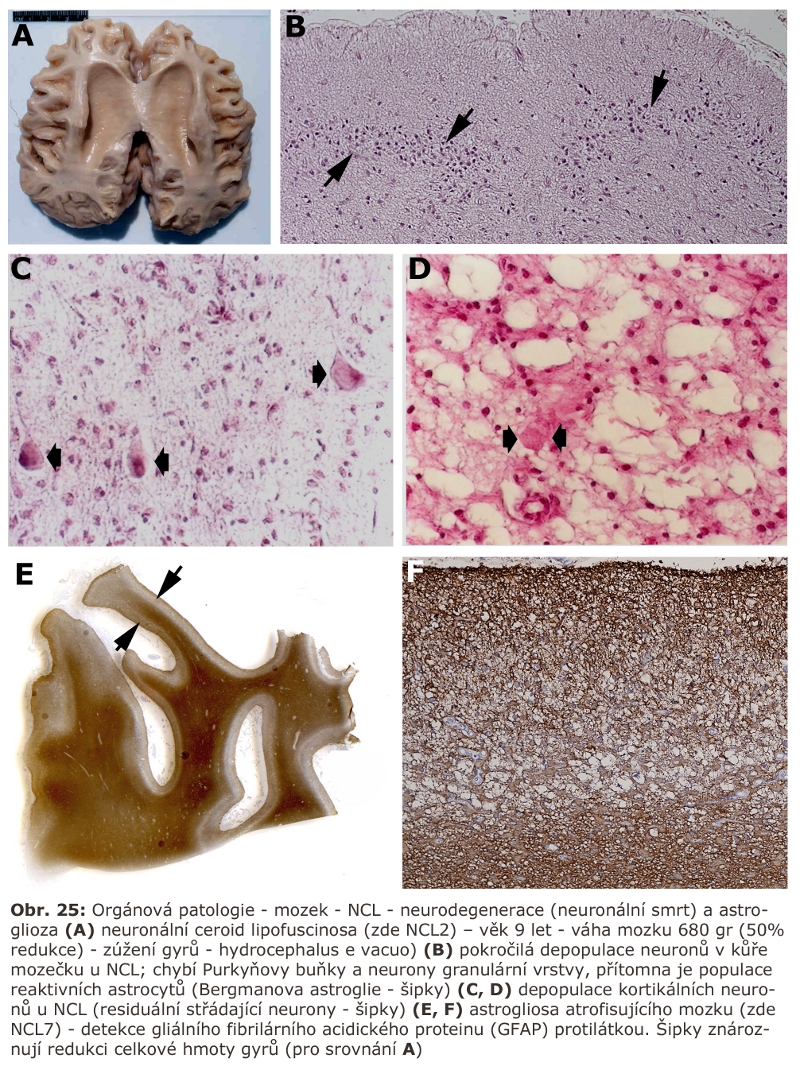
U některých lysosomálnch enzymopatií dochází ke střádání i v arachnotelu leptomeninx (Obr. 26).
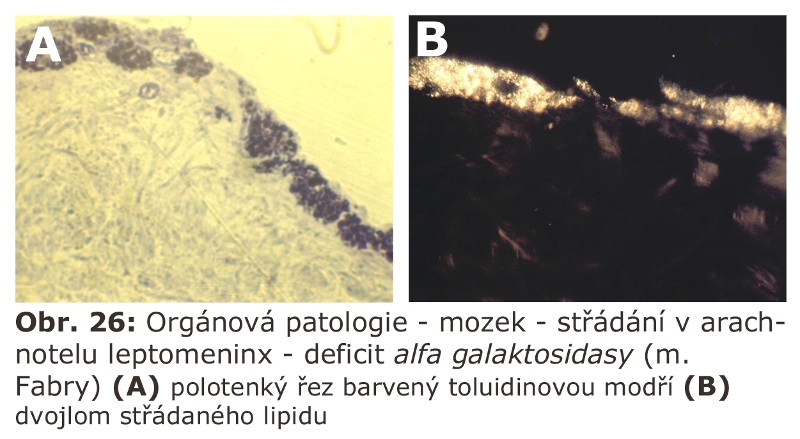
Střádání v oligodendroglii. Jde o dvě jednotky: metachromatickou leukodystrofii (sulfatidosu, deficit arylsulfatasy A) a Krabbeho nemoc (deficit β-galaktosidasy).
Situace je zde velmi odlišná od lysosomálního střádání v ostatních buňkách organismu. Důvod je ten, že jde o poruchu obratu komplexu myelinových membrán, které jsou součástí oligodendroglie samotné a kterými oligodendroglie obkružuje axon (Obr. 27). Jakým způsobem je realisován obrat lipidních komponent (galaktocerebrosidu a sulfatidu) tohoto komplexního systému membrán není zcela jasné. Tím se oligodendroglie liší od všech ostatních buněčných typů, které mají na periferii jednoduchou buněčnou membránu, která komunikuje s extracelulárním prostorem procesem endocytosy, případně fagocytosy (viz shora).
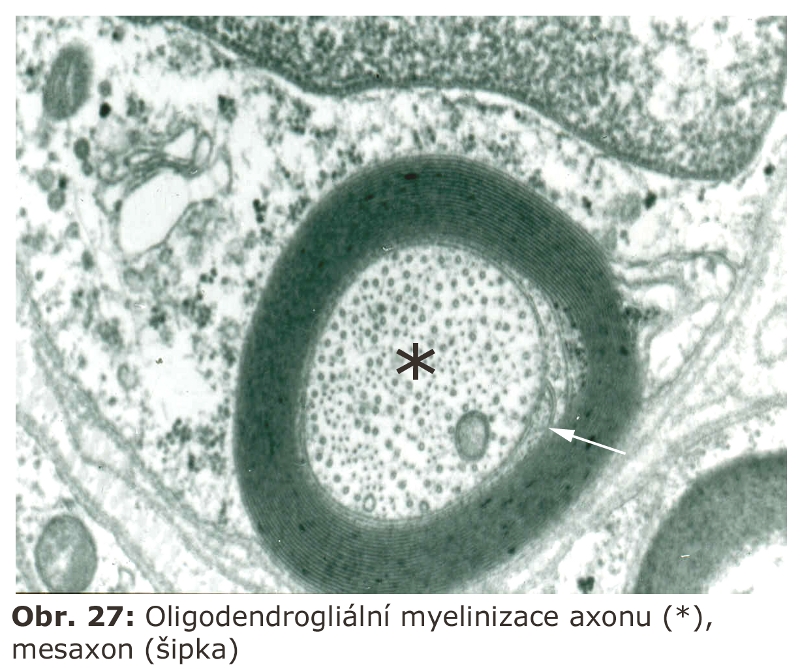
Klasická akumulace střádaného lipidu v lysosomech je vyjádřená minimálně, v mnoha případech chybí. U Krabbeho nemoci se předpokládá toxický vliv lysoderivátu galaktocerebrosidu (údajně syntetického původu), který vede k zániku oligodendroglie již v počátečním stadiu střádání. Sekvence změn u Krabbeho nemoci je na Schématu 13.

V případě sulfatidosy se předpokládá abnormální zvýšení koncentrace sulfatidu v lamelách myelinu, což snižuje jeho stabilitu. Situace na úrovni molekulární patologie buňky není doposud zcela vysvětlena. Názory na mechanimus změn, zodpovědných za vznik leukodystrofie u sulfatidosy, jsou shrnuty na Schématu 14 (pro srovnání progrese změn u m. Krabbe Schéma 13)
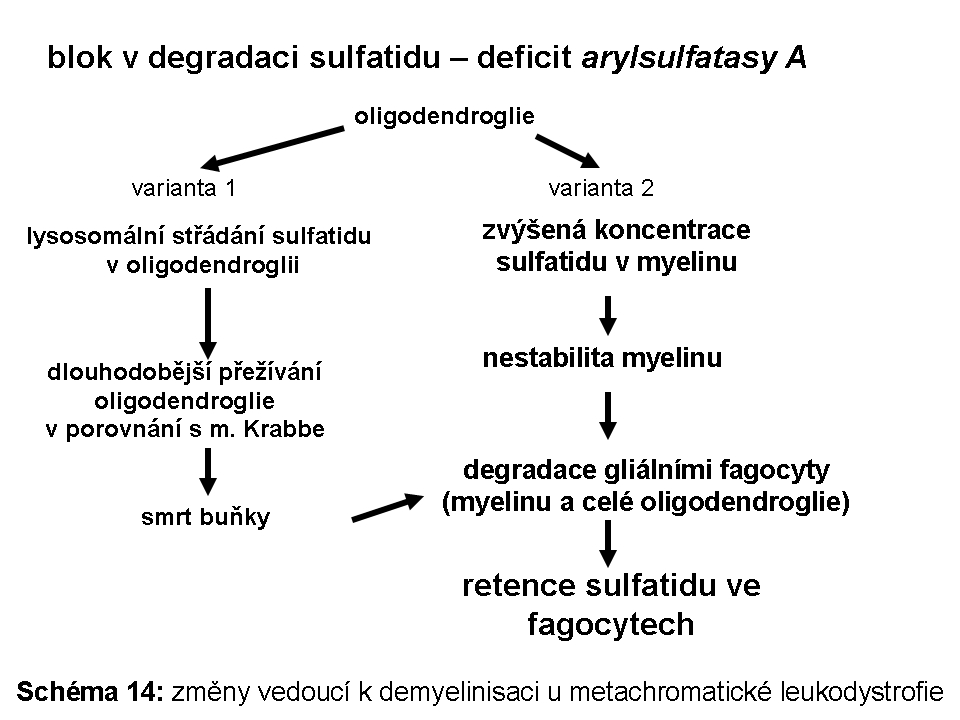
Dá se shrnout, že střádání vede k zániku oligodendroglie, tím k nestabilitě myelinu (který je součástí oligodendroglie) a k jeho odbourání. Následkem je lysosomální leukodystrofie. Gliové fagocyty degradují všechny myelinové komponenty s výjimkou substrátu deficitního enzymu. Proces střádání se tedy projevuje v plné intensitě sekundárně až v gliálních fagocytech, které u Krabbeho nemoci představují jediný střádací buněčný typ (Obr. 28 A) U sulfatidosy jsou střádáním postiženy i astrocyty (Obr. 28 B) a některé neurony.
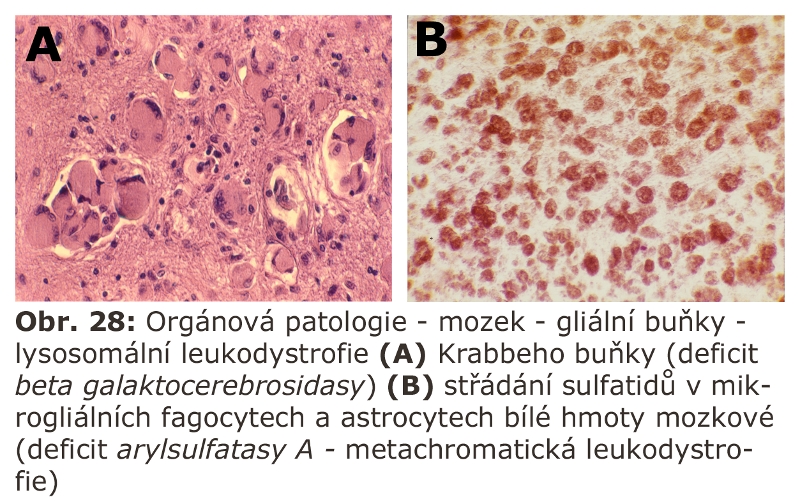
Postižení sítnice (jedná se vlastně o jedinou část CNS dostupnou zevnímu vyšetření) se projevuje abnormálním žlutavým zabarvením, které je způsobeno akumulovaným střádaným lipidem v neuronálních perikaryích. Od něj se odráží tmavší, červená barva foveoly, kde perikarya chybí. Tento abnormální obraz je podstatou tzv. třešňové skvrny na očním pozadí popsané již na konci 19. století u prvých případů Tay-Sachsovy nemoci. K tomuto projevu dochází typicky u gangliosidos (GM2, GM1), vzácně u jiných lipidos, obecně však u stavů s vyznačeným střádáním lipidu v neuronech. Je-li menší intensita střádání, žlutá barva se koncentruje do perifoveolární oblasti, kde je koncentrace neuronů nejvyšší.
Další variantou postižení je tzv. pigmentová retinitida, jejíž podstatou je atrofie sítnice daná degenerací a výpadky neuronů, gliosou, postižením retinálního pigmentového epitelu, jeho rozpadem, fagocytosou melaninu makrofágy a jejich migrací sítnicí. Souhrnně pak tyto změny dávají vznik tzv. "pigmentové retinitidě", což není nic jiného než ztráta integrity vrstvy retinálního epitelu a disperse fagocytujících melanofágů (Obr. 29)
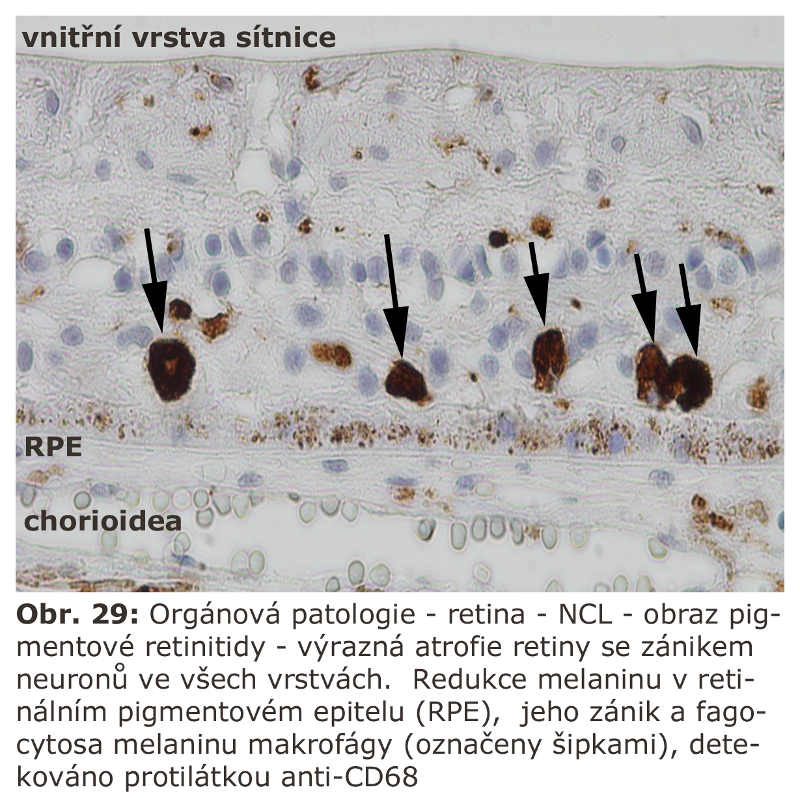
Pokud se střádání manifestuje v makrofázích, dochází k výraznému zvětšení sleziny, lymfatických uzlin, ale i jater. Střádající makrofágy mohou výrazným způsobem infiltrovat plíce. Při splenomegalii dochází až k dysfunkci sleziny (hypersplenismu), jejímž projevem je dřeňový útlum. Postižením makrofágů se vyznačuje Gaucherova nemoc (deficit β-glukocerebrosidasy) a m. Niemann-Pick, typy A a B (deficit kyselé sfingomyelinasy) (Obr. 30).
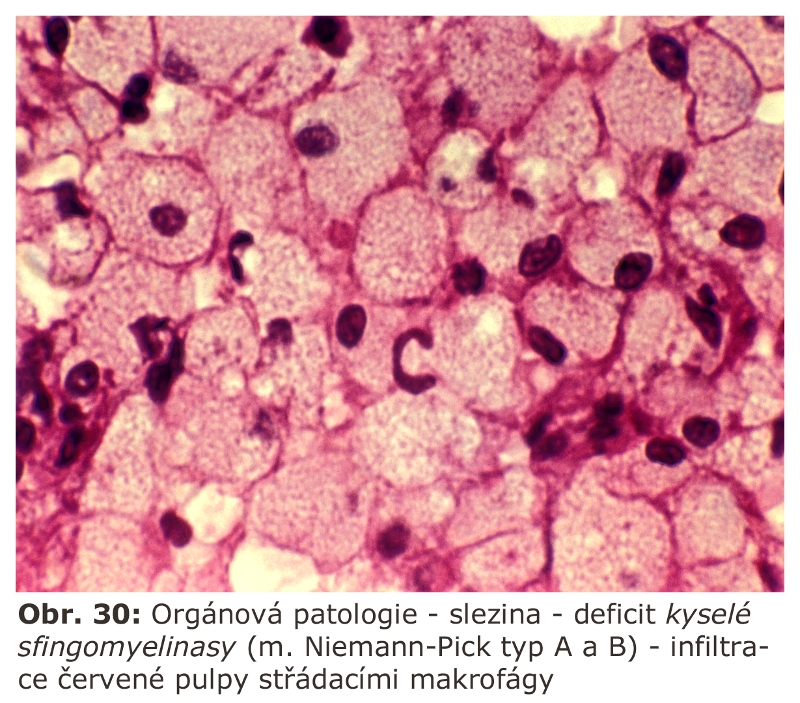
Střádání v periferních leukocytech je velmi dobře známo z cytologické analysy periferních krevních nátěrů, zejména o glykoproteinos, typicky u α-manosidosy (Obr. 31 A), u řady mukopolysacharidos (Obr. 31 E), u mukolipidosy II (Obr. 31 C, D) a u neuronální ceroid lipofuscinosy typu 3 (NCL3) (Obr. 31 F). O bližší povaze tzv. Alder-Reilyho granul, které jsou též vyjádřením střádání v leukocytech a zejména o jejich vztahu k systému LRO (viz níže), není známo nic podstatného. Jsou definovány jako střádací vakuoly s densním centrem.
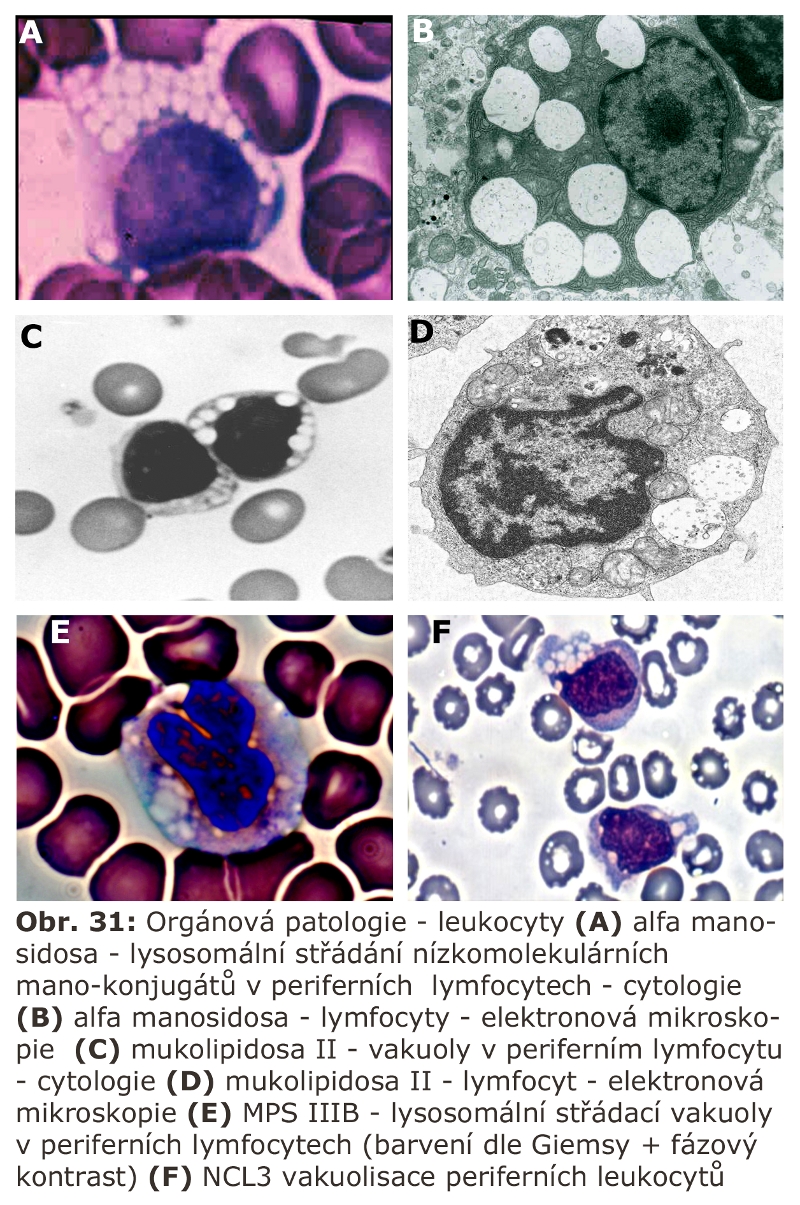
Při střádání v krevních kapilárách dochází velmi často k vzniku tzv. angiokeratomů. Angiokeratomy se běžně nalézají u Fabryho nemoci (deficit α-galaktosidasy), u které mají na kůži svá predilekční místa. Jedním z nich je oblast okolo pupku (Obr. 32 A). Jejich podstatou jsou fokální distense konečných úseků kožních kapilár postižených střádáním, kolem těchto distensí se vytváří "límec" proliferujících keratinocytů (Obr. 32 B). Angiokeratomy se vyskytují i u Schindlerovy nemoci (deficit α-N-Acetyl-galaktosaminidasy), u α-fukosidosy a β-manosidosy. Všem je společné významné střádání v cévním endotelu. Mechanismus jejich vzniku není jasný. Jsou pozoruhodné diskrepancí mezi postižením cévy a minimálním stupněm střádání. Nevyskytují se mimo kůži a sliznice.

Postižení cévní stěny (arterií i žil) je nejvíce prostudováno u Fabryho nemoci. Vedle endotelií jsou masivně postiženy střádáním hladké svalové buňky (Obr. 33). To vysvětluje cévní varikosity, přímo patrné v sítnici a dále celý komplex arteriálních problémů u této enzymopatie, od narušené funkce (tendence ke spasmům) až po fibrosu, kalcifikace a vznik aneurysmat.
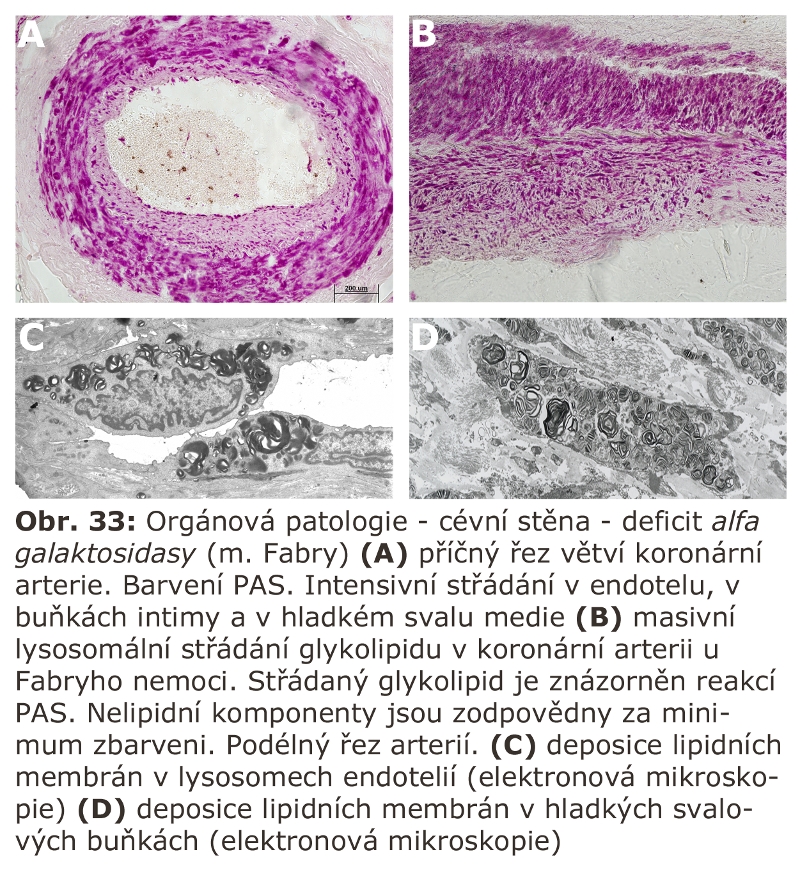
Postižení kůže je známo zejména postižením cévního endotelu (viz výše). Významné postižení je známo u potních (ekrinních) žlázek, typicky u mukopolysacharidos (Obr. 34) a ve skupině neuronálních ceroid lipofuscinos (Obr. 35). Zejména ve druhé jmenované skupině je kožní biopsie resp. potní žlázky v ní obsažené důležitým diagnostickým vzorkem. Málo sledovaná je epidermis, i když patří též k buňkám postiženým lysosomálním střádáním. Známé je postižení epidermis u některých mukopolysacharidos a u střádání sialové kyseliny (mutace membránového transportéru - Obr. 36). V lysosomech takto postižených keratinocytů se nacházejí i normálně fagocytované melanosomy. Jejich degradace je zvýšená, což vysvětluje tendenci k výrazné ztrátě pigmentace kůže u těchto patologických stavů.
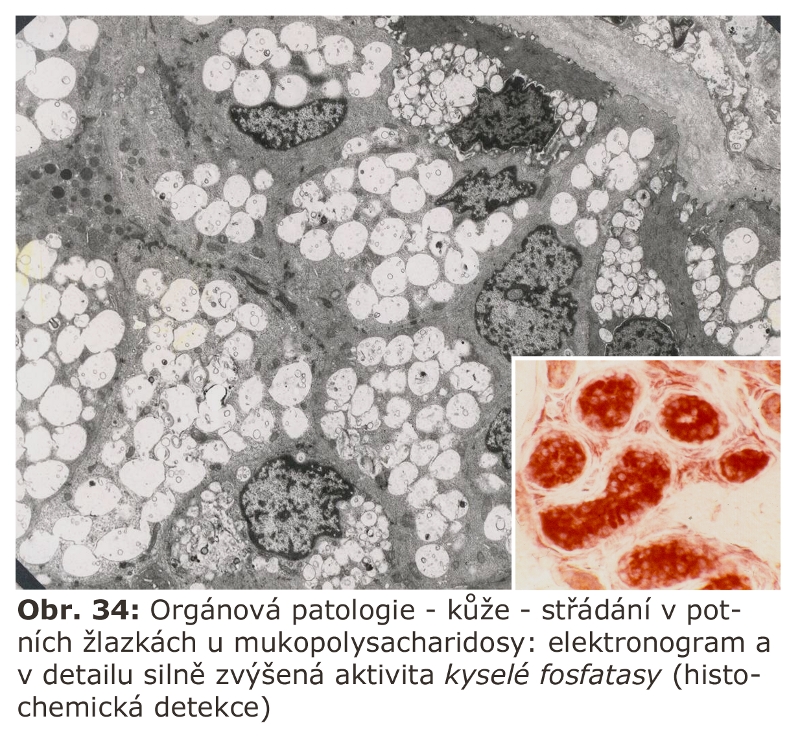
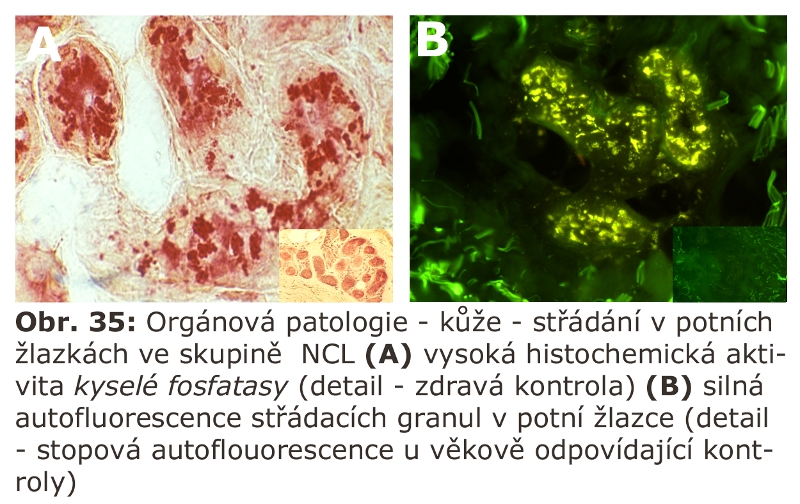
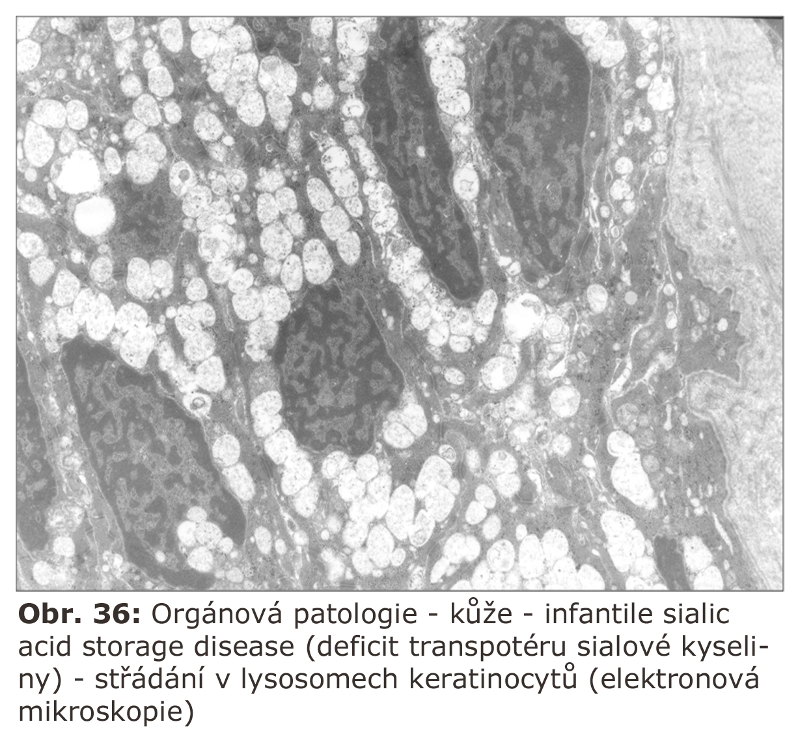
Plíce jsou nejvýrazněji postiženy u střádacích onemocnění postihující makrofágy (m. Niemann-Pick A,B, vzácněji C; m. Gaucher). Postižení je různě vyjádřeno, někdy je tak intensivní, že vede až k respirační insuficienci. Důvody, proč se v některých případech střádací makrofágy akumulují v plicích (jde převážně o alveolární lokalisaci, i když jejich přítomnost v alveolárních septech byla opakovaně popsána) nejsou jasné. Střádání může být dále vyjádřeno v rámci postižení hladkého svalu cévního (např. u Fabryho nemoci) či v rámci postižení periferního nervového systému. Střádání v respiračním epitelu je známé u některých lysosomálních poruch jako je deficit kyselé sfingomyelinasy (Obr. 37 A), ale nebylo systematicky sledováno. Časté je u Fabryho nemoci (Obr. 37 B).
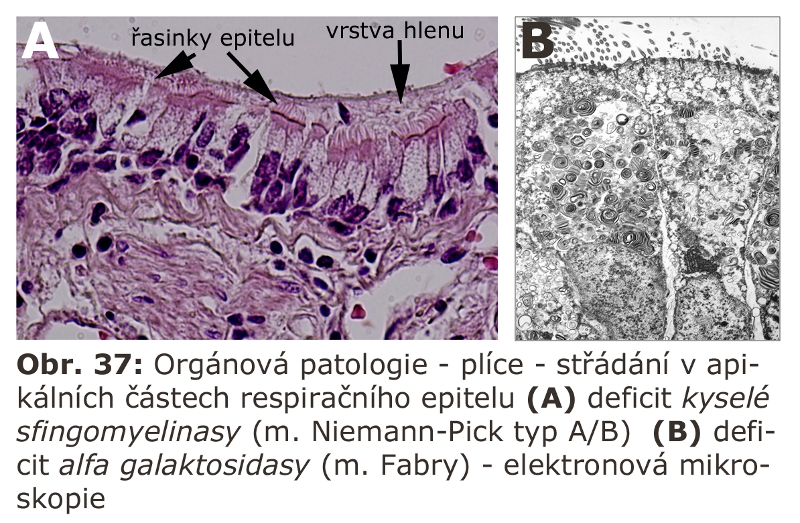
Kosterní sval je velmi výrazně postižen u deficitu α-glukosidasy (m. Pompe - glycogen storage disease type II). Výsledkem je myopatie. Ta může být hlavním příznakem u dospělé varianty Pompeho nemoci (deficit lysosomální α-glukosidasy). Změny odpovídají lysosomální akumulaci glykogenu, které mohou být vyjádřeny v různé intensitě v jednotlivých vláknech (Obr. 38 A). Manifestace ve svalu je unikátní tím, že je výrazně aktivována autofagie, které přispívá podstatným způsobem k postižení svalu (Obr. 38 C, D).
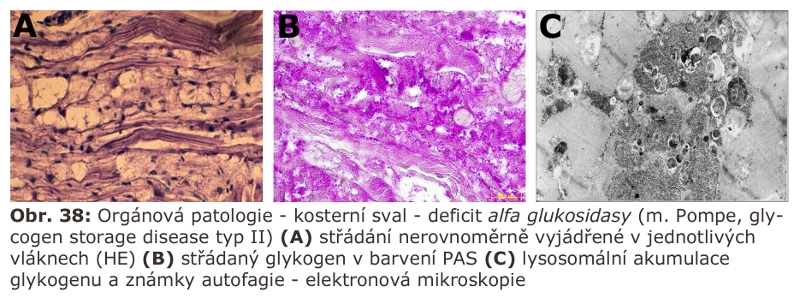
Nízké pH lysosomů odpovídá za fenomén distense endosomů, způsobený aplikací slabých organických bází. Tyto jsou při extracelulárním pH elektro neutrální a procházejí volně membránami buněk do různých intracelulárních oddílů buňky. V kyselém kompartmentu (terminální části Golgiho aparátu a ELS) addují proton a pro takto vzniklé ionty přestává být membrána kompartmentu prostupná. Osmoticky je nasávána voda za současné distense lysosomů.
Je velmi dobře známo, že značná část látek, podávaných nebo testovaných jako léky, může vyvolat obraz identický s lysosomální enzymopatií. Společným jmenovatelem celé řady těchto látek je jejich amfifilní povaha (hydrofobní i hydrofilní zároveň) a přítomnost dusíkového atomu, který se může protonací změnit na kvarterní amoniovou bázi. Tyto látky mají schopnost vázat lipidy, zejména fosfolipidy řady fosfoglyceridů, a vytvářet s nimi komplexy, které jsou těžko štěpitelné příslušnými enzymy. Všechny tyto látky zároveň zvyšují endosomální a lysosomální pH, čímž celkově přispívají ke snížení hydrolytické kapacity lysosomálních enzymů.
Nejznámější příklady zahrnují:hexestrol (diethylaminoethoxyhexestrol), původně podávaný jako koronární vazodilatans, vyvolává při delším podávání obraz generalisovaného střádání fosfolipidů. Masivní střádání fosfolipidů v řadě buněk byl vysvětlován inhibicí fosfolipasy A1. Proces je fenotypově v mnoha rysech velmi blízký m. Niemann-Pick typ A a B (deficit kyselé sfingomyelinasy). Zvláště masivně jsou postižena játra (Obr. 39 A, B)
gentamicin (aminoglykosidové antibiotikum) je známý zejména svým nefrotoxickým působením. V ledvinách, ale i jinde, vyvolává výrazné lysosomální střádání fosfolipidů.
chlorochin (antimalarikum) vyvolává lysosomální střádání v nejrůznějších orgánech. Paralelně inhibuje celou řadu enzymů, včetně kyselé sfingomyelinasy.
tricyklická antidepresiva (imipramin, clomipramin, iprindol) vyvolávají v experimentu obraz generalisované lipidosy. Podobně se chová anorektikum chlorfentermin.
Některé z těchto látek se váží přímo na aktivní centra enzymů a působí jako enzymové inhibitory. Nejznámějším takovým inhibitorem je v současné době konduritol β-epoxid, specificky inhibující lysosomální β-glukocerebrosidasu.
Tiloron a Suramin indukují změny typu mukopolysacharidosy. Tiloron inhibuje degradaci dermatan sulfátu. Výsledkem je obraz srovnatelný s mukopolysacharidosou (Obr 39 C).
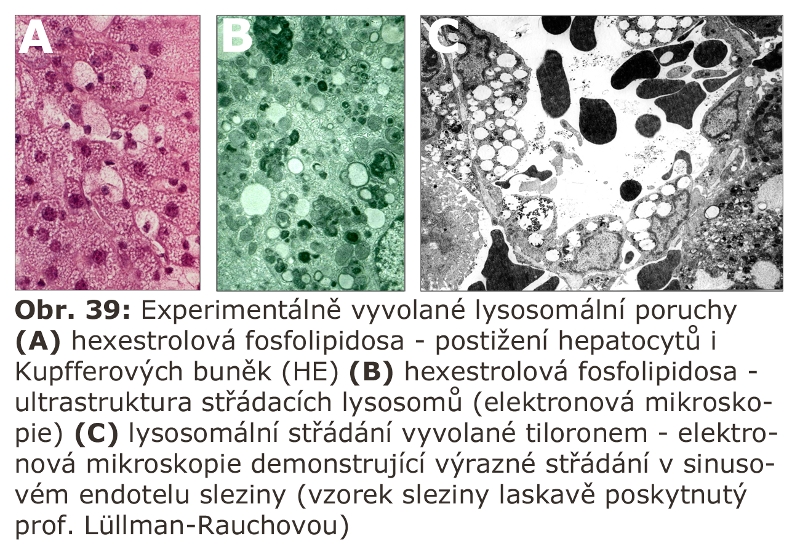
Detekce lysosomů vychází ze znalosti základů jejich strukturní biologie.
Detekce lysosomálních luminálních komponent (Schéma 15)Klasickou cestou je průkaz aktivity lysosomálních enzymů, což vyžaduje tkáň nenarušenou fixací. Tato problematika je natolik známa, že bližší zmínka v tomto textu je nadbytečná. Jen okrajově je vhodná poznámka, že použití Gomoriho techniky průkazu kyselé fosfatasy vedlo k identifikaci lysosomů na ultrastrukturální úrovni (Obr. 40 D).
Metodou pro běžnou aplikaci v dostupných rutinních tkáňových vzorcích (obvykle formolem fixované a do parafínu zalité tkáňové vzorky) je imunohistochemická nebo imunofluorescenční detekce katepsinu D (ubikviterní lysosomální proteasa). Antigenní epitop katepsinu D je detekovatelný i po aldehydové fixaci, odvodnění a zalití do parafinu (Obr. 40 A). V kryostatových řezech (v nátěrech nebo monolayerech kultivovaných buněk), může být na obtíž solubilita katepsinu D (jde o solubilní intralysosomální hydrolasu). Pak je na místě testovat fixaci metanolem. Imunohistochemické přístupy mohou být použity i pro detekci jiných intralysosomálních komponent, např. saposinů či lysosomálních enzymů.
Detekce lysosomálních membránových komponent.Komerčně jsou dostupné protilátky proti LAMP1, LAMP2, CD63 a LIMP2. Jde o integrální membránové glykoproteiny. Nehrozí jejich solubilita. Persistují i po standardním zpracování vzorků, takže lze detekci provést i v parafinových řezech (Obr. 40 B, C). Pro detekci makrofágů je běžně používána monoklonální protilátka CD68, která detekuje lysosomy makrofágů.
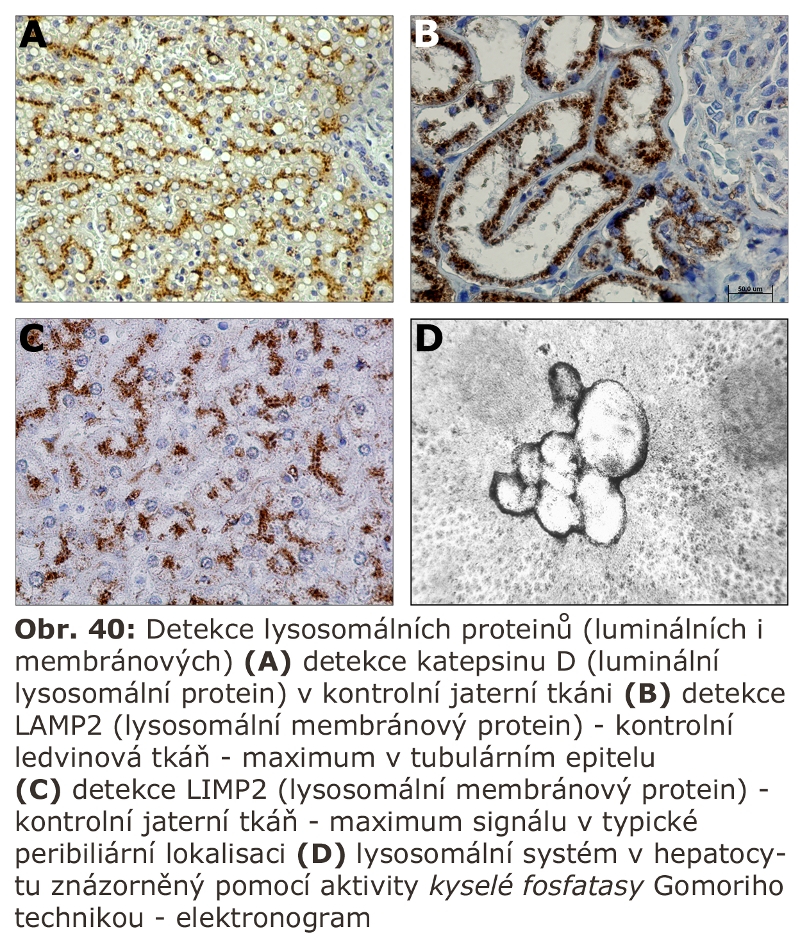
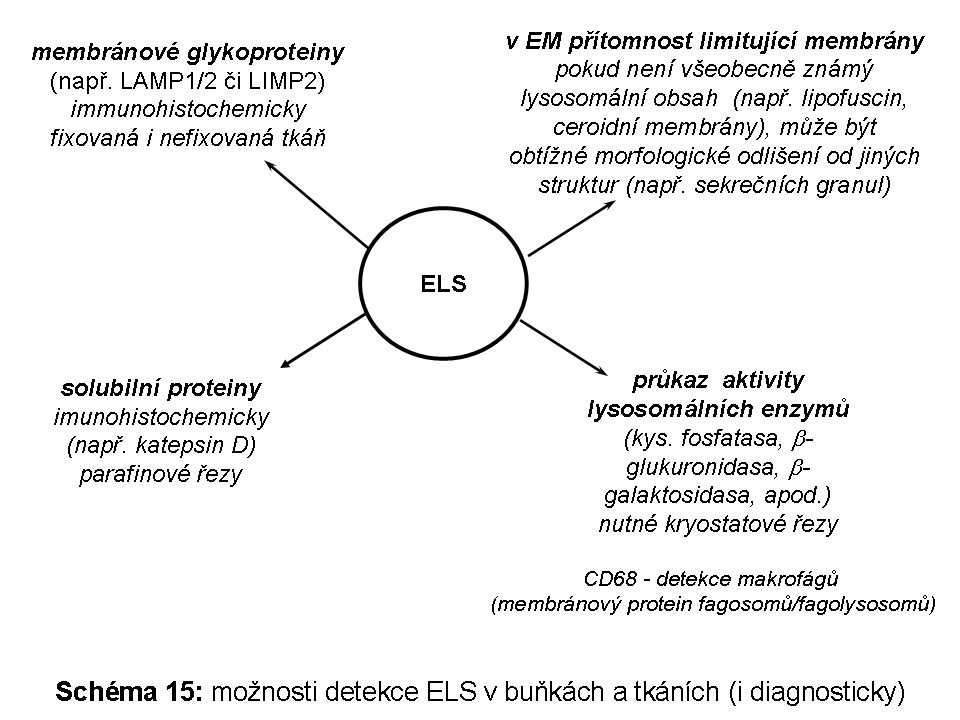
Klasické biochemické in vitro techniky, testující aktivitu lysosomálních enzymů, mají své diagnosticky nezastupitelné místo, ale v některých případech jsou zcela nezbytné další metodické přístupy. Ukazuje se, že aktivita enzymu in vitro je sice stanovena naprosto exaktně, ale po stránce biologické nemusí odpovídat situaci in vivo. Jednou z příčin je, že se většinou nepoužívá přirozený, ale syntetický substrát, jehož vlastnosti jsou odlišné. Korelace takto změřené residuální aktivity lysosomálního enzymu u lysosomálních enzymopatií neskýtá možnost zcela spolehlivé korelace s klinickou mírou onemocnění.
Stanovit kritickou dolní hranici reziduální aktivity, která by vedla k rozvoji střádání a tím klinického onemocnění, je rovněž velmi obtížné. Lze pozorovat rychlý a smrtelný průběh onemocnění při hodnotách residuální enzymové aktivity z rutinního měření v řádu 10-20% normy, na druhé straně lze při podstatně nižších aktivitách pozorovat velmi příznivý a mírný fenotyp, případně průběh zcela klinicky asymptomatický. Krajním příkladem je tzv. pseudodeficience, kdy při residuálních hodnotách 10-20% normální aktivity chybí nejen klinické příznaky, ale i jakékoliv známky střádání na buněčné úrovni.
Velmi cennou pomocí pro řešení těchto situací jsou takzvané dynamické zátěžové testy (loading testy) lysosomálních enzymů, prováděné v buněčných kulturách mutantních buněk (např. fibroblastů, EB transformovaných B lymfocytů či buněk trofoblastu).
Principem je využití endocytosy značeného kritického substrátu (přirozený substrát značený fluorogenem, radioisotopem nebo stabilním isotopem), podaného buď volně ve formě micel nebo prostřednictvím receptorem zprostředkované endocytosy, která je mnohonásobně efektivnější než endocytosa vodné fáze (fluid phase). Substrát je ve druhém případě aplikován ve formě liposomu obsahujícího ligand pro buněčný povrchový receptor (např. radioaktivně značený sfingomyelin zabudovaný do liposomu s apoB nebo přímo do LDL při diagnostice deficitu kyselé sfingomyelinasy - m. Niemann-Pick typ A a B).
Uspořádání testu umožňuje posuzovat dynamiku degradace endocytovaného lipidu a korelovat tyto hodnoty s hodnotami změřenými in vitro. Hodnoty těchto zátěžových testů jsou mnohem "fysiologičtější" a jejich korelace s klinickým fenotypem je dokonalejší. Např. u shora zmíněné pseudodeficience jsou hodnoty zátěžového testu v normálních mezích. Znamená to, že podmínky pro normální aktivitu enzymu s pseudodeficientní alelou jsou zajištěny pouze v podmínkách in vivo. Totéž platí pro případy deficitu proteinových aktivátorů sfingolipidových hydrolas (saposinů), které se nedají rutinní enzymologií in vitro prokázat, ačkoliv reakce neprobíhá a substrát se hromadí stejně jako v případě mutovaného enzymového proteinu.
Podobně jako u jiných monogenně dědičných onemocnění je znalost genu(ů) odpovědných v případě patogenní variace (mutace) v jejich sekvenci za rozvoj fenotypu onemocnění naprosto zásadní nejen pro molekulárně genetickou diagnostiku v postižených rodinách, ale i pro další výzkum příslušného onemocnění (např. možnost vytváření modelových organismů, krystalisace proteinu pro zjištění terciární nebo kvarterní struktury apod.). Jak bylo uvedeno v úvodu tohoto textu, u většiny lysosomálních enzymopatií byla nejdříve stanovena deficitní enzymová aktivita a až následně byl nalezen gen kódující příslušný enzym. Tato skutečnost, tedy apriorní znalost deficitní aktivity, vždy znamenala ulehčení identifikace kritického genu. Komplikovanější byla (je) situace u neenzymatických deficitů, kde bylo (je) i přes použití více či méně sofistikovaných algoritmů vazebné analysy obtížnější prokázat kauzalitu vztahu mezi nově nalezenými mutacemi v dosud s chorobou neasociovaných genech a rozvojem klinického fenotypu.
Molekulárně genetická diagnostika představuje vrchol diagnostického algoritmu u každé dědičné monogenní choroby a v případě, kdy se nedaří mutaci prokázat, by neměl být příslušný případ uzavírán i při positivitě jiných diagnostických kriterií (např. enzymatický deficit). Vždy se může jednat o vliv nějakého doposud neidentifikovaného modifikujícího genu. Obrovskou výhodou molekulárně genetické diagnostiky je možnost provádět ji s vysokou přesností prenatálně, a i proto je nutné v rodinách, kde se onemocnění vyskytlo, mutace stanovovat, i když není možné v současnosti nabídnout kauzální léčbu žijícím pacientům. Dále je potřeba uvést, že určité typy terapie (např. chaperonová, viz níže) budou vždy vhodné pouze pro pacienty s určitým, předem známým a testovaným typem mutace. V konkrétním případě této modality léčby lze positivní terapeutický efekt očekávat pouze u pacientů postižených mutací se zachovanou residuální aktivitou a positivní odpovědí na stabilisaci mutantního konformeru.
Jak vyplývá z výše uvedeného, skupina lysosomálních střádacích onemocnění představuje soubor nosologických jednotek, které přestože celou řadu klinických, buněčně biologických, buněčně patologických a molekulárně patologických aspektů vzájemně sdílejí, jsou jako celek značně heterogenní. Se zřetelem k této skutečnosti je třeba přistupovat ke studiu jejich patogeneze, ale také k experimentálním a následně i klinickým pokusům o jejich terapii.
Oblast výzkumu a vývoje terapeutických přístupů v oblasti lysosomálních střádacích onemocnění má, podobně jako u jiných vzácných metabolických onemocnění, svá specifika, která musí být nutně zvážena a odpovídajícím způsobem řešena, pokud má být vůbec uvažováno o vývoji a implementaci jakýchkoliv kauzálních terapeutických postupů. Mezi taková specifika patří zejména následující body:
Z biologického pohledu a při současné kritické znalosti mechanismů participujících na patogenesi lysosomálních střádacích onemocnění (viz výše) je možné o obecných přístupech k terapii a směrech jejího dalšího výzkumného rozvoje uvažovat v následujících rovinách, tzn. pokusit se v experimentální a následně i v klinické terapii o následující:
Aktuální skutečností je tendence kombinovat jednotlivé uvedené přístupy a vytvářet efektivnější biologicky opodstatněné aditivně fungující kombinované terapeutické protokoly.
Ad. 1Terapie redukcí syntesy substrátů (substrate reduction therapy - SRT). Tento přístup je v současnosti, v podstatě výlučně, spojován s klinickým terapeutickým použitím inhibitoru syntesy glykosfingolipidů - N-butyl-deoxyjonojirimycinem (NB-DNJ). Snížení syntesy glykosfingolipidů může znamenat snížení nároků na jejich následnou degradaci a positivni efekt lze tedy očekávat u těch lysosomálních střádacích onemocnění, která jsou spojena s jejich primární nebo sekundární akumulací. V případě NB-DNJ se tedy o jedná látku s konkrétním terapeutickým využitím u m. Gaucher (deficit β-glukocerebrosidasy) nebo m. Niemann-Pick typ C, kde v některých buněčných typech (zejména neuronech) dominuje z neznámého důvodu střádání právě glykosfingolipidů nad střádáním neesterifikovaného cholesterolu.
Ad. 3S odkazem na dříve uvedenou část týkající se funkčních dopadů lysosomálního střádání na fysiologii buňky je vhodné zmínit několik potencionálně přínosných terapeutických přístupů, které přímo odráží výsledky základního buněčně biologického výzkumu v této oblasti.
Konkrétně se v současnosti testují možnosti rekonstituce kalciové intracelulární rovnováhy porušené v souvislosti s lysosomálním střádáním použitím látek na bázi kurkuminu. Skutečným hitem posledních let je, na zvířecích modelech provedený, průkaz positivního vlivu 2-hydroxypropyl-β-cyklodextrinu na redistribuci neesterifikovaného cholesterolu u m. Niemann-Pick typ C. Z dalších je pak možné uvést pokusy o terapeutické použití antioxidantů (např. N-Acetyl-cystein) pro zlepšení klinického fenotypu u některých lysomálních střádacích onemocnění. Také se testuje použití protizánětlivých léků. Experimentálním podkladem použití těchto léčiv je skutečnost, že jedním z hlavních obecných efektů lysosomálního střádání v CNS je potenciálně poškozující aktivace mikroglie, což lze, ve specifickém smyslu slova, považovat za zánětlivou reakci.
Ad. 4Symptomatická léčba ve všech klinických oborech, jichž se patologie lysosomálního střádání dotýká. Pro definování takové oborové šíře je možné se podržet výše uvedené části "orgánová patologie", nebo v závěru uvedeného stručného přehledu specialisací původních autorů popisů jednotlivých lysosomálních střádacích onemocnění (viz níže). V podstatě neexistuje obor lidské medicíny (Schéma 16), jehož specialisace by nezahrnovala symptomatologii spojenou s nějakou formou lysosomálního střádání.

B. J. Underwood et al.: Autophagy and Human Genetic Disease. (Metabolic & Molecular Bases of Inherited Disease A. Valle et al., Eds. McGraw Hill, New York, 2007)
M. Eckhard: Pathology and current treatment of neurodegenerative sphingolipidoses. (Neuromolecular Med. 12: 362-382, 2010)
B. Levine, G. Kroemer:Autophagy in the pathogenesis of disease. (Cell 132: 27-42, 2008)
D. Glick et al.: Autophagy: cellular and molecular mechanisms. ( J. Pathol. 221: 3-12, 2010)
G. J. Doherty, H.T. McMahon: Mechanisms of endocytosis. ( Ann. Rev. Biochem. 78: 857-902, 2009)
E. B. Vitner et al.: Common and uncommon pathogenic cascades in lysosomal storage diseases. ( J. Biol. Chem. 285: 20423-20427, 2010)
S. U. Walkley: Pathogenic cascades in lysosomal disease - Why so complex? ( J. Inherit. Metab. Dis. 32: 181-189, 2009)
Jednou z velmi častých nespecifických poruch lysosomálního aparátu je vznik tzv. residuálních tělísek. Patří mezi ně skupina tzv. lipopigmentů a residuální tělíska obsahující kovy (sole železa a mědi).
Lipopigment je historický termín (stále méně používaný) pro tato lysosomální residuální tělíska. Naznačuje dlouho převládající předpoklad, že jde o pigmentované látky, derivované z lipidů jejich peroxidací, což bylo v řadě situací vyloučeno. V literatuře jsou, dnes již historicky, zakotveny dvě formy (varianty) lipopigmentu: ceroid a lipofuscin. Po určitou dobu bylo používáno dělení dle Pearse, podle kterého vznikají lipopigmenty peroxidací nenasycených lipidů, při čemž ceroid a lipofuscin byly definovány jako různě pokročilá stadia lipoperoxidace (ceroid iniciální, lipofuscin pokročilejší). Gedigk a Totovič odlišovali obě varianty lipopigmentu podle mechanismu vzniku: ceroid v procesu fagocytosy, lipofuscin v procesu autofagocytosy. V současnosti se konsensuálně rozlišují oba typy (Schéma 17). Jde o formální rozlišení, které je nepochybně dočasné.
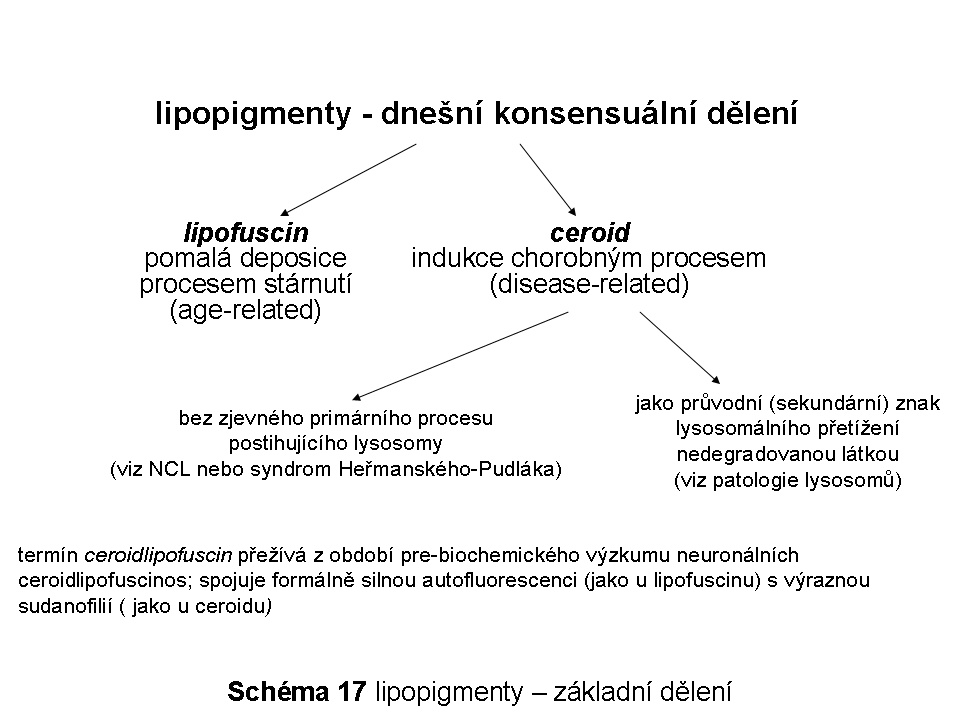
4.9.1.1. Ceroid (odvozeno od řeckého slova žlutý) je tedy podle současné definice lipopigment indukovaný patologickým procesem. Vyznačuje se v některých případech nahnědlou barvou. Je silně hydrofobní, barví se tedy silně nepolárními histologickými barvivy (tuková červeň, sudanová čerň), není rozpustný v běžných tukových rozpustidlech (dá se prokázat i v parafinu) a žlutě fluoreskuje po excitaci světlem o vlnové délce 360-400 nm.
V nejklasičtější formě se ceroid vyskytuje v histiocytech, jejichž lysosomální systém byl přetížen dlouhodobou exogenní náloží (fagocytovanými lipidními látkami nebo buňkami bohatými na strukturální lipidy jako jsou fragmenty myelinu, lipoproteiny, krevní destičky a další. Histiocyty přeplněné ceroidními granuly mají v klasických hematologických cytologických barveních velmi impresivní obraz masivní granulace modré nebo modrozelené barvy (mořsky modré - sea blue), která jim dala jméno sea-blue histiocyty (Obr. 41). Ceroid může být jedinou komponentou histiocytárních lysosomů. Typickým příkladem je excesivní histiocytární ceroidosa u syndromu Heřmanského a Pudláka a Chediak-Higashiho syndromu (viz níže). Běžně se však takové histiocyty bohaté na ceroid vyskytují u nejrůznějších lysosomálních enzymopatií, kdy se ceroid akumuluje sekundárně jako jeden z důsledků lysosomální dysfunkce. Typickym příkladem je protrahovaná forma Niemann-Pickova onemocnění (deficit kyselé sfingomyelinasy - Obr. 42) V takových případech je možné vždy histochemicky prokázat v lysosomálních vakuolách, doposud nezaplněných ceroidem, akumulovaný neštěpený enzymový substrát (např. lipid), který je primárním projevem poruchy, ať již geneticky podmíněné, nebo získané.
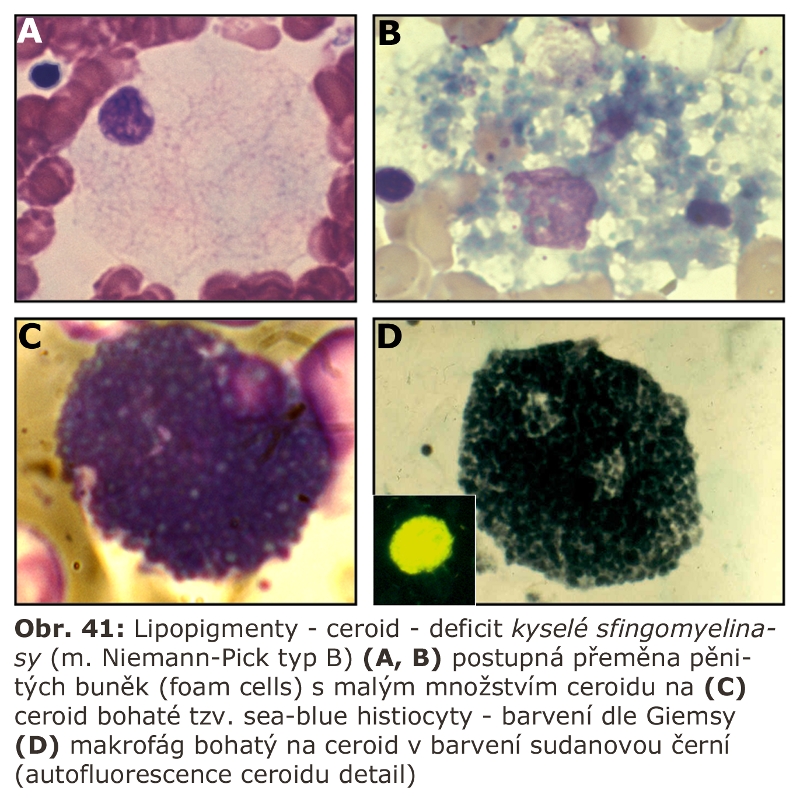
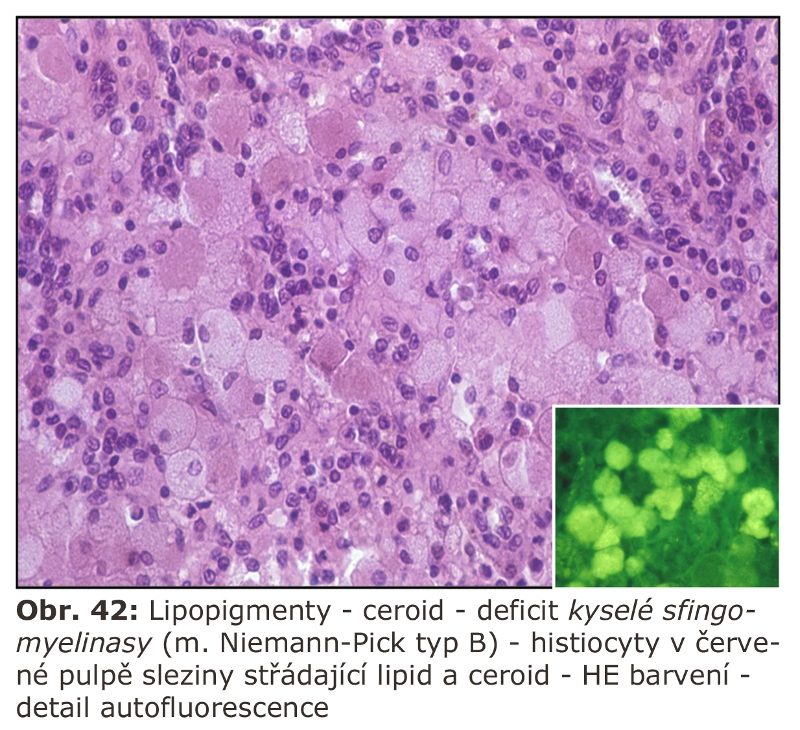
K indukci ceroidu dochází často v lysosomech jiných buněčných typů, postižených lysosomálním střádáním, zejména, jde-li o pomalu probíhající formy. Velmi často je tomu v neuronech u gangliosidos (Schéma 19) a u mukopolysacharidos, nebo obecně v lysosomech celé řady buněčných typů. S tímto faktem nutno počítat při analyse tkání u celé řady dalších lysosomálních poruch. Je to jen jeden z výrazů sekundární dysfunkce přetíženého ELS (viz výše).
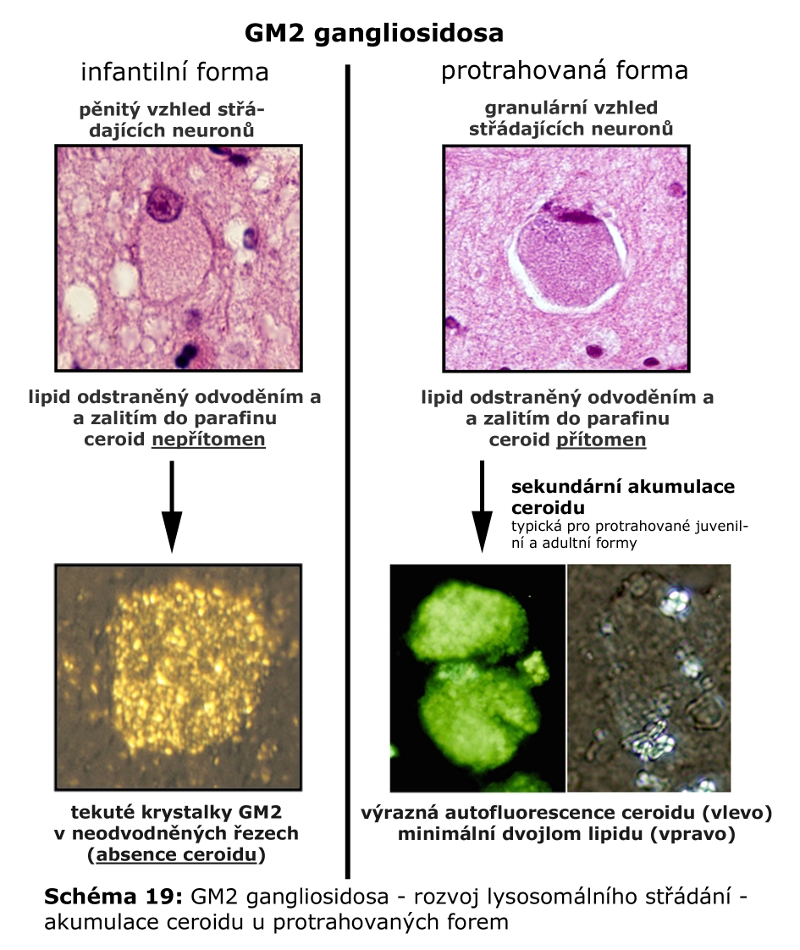
Jako příklad uvádíme Fabryho nemoc, pomalu progredující lysosomální enzymopatii (defekt α-galaktosidasy, střádání globotriaosylceramidu). Indukci ceroidu lze prokázat ve střádacích kardiomyocytech srovnáním dvojlomu (střádaný lipid) s autofluorescencí (Obr. 43 A, B), nebo srovnáním imunodetekce lipidu s autofluorescencí, která ukáže ceroid jako hlavní komponentu střádacího lysosomu (Obr. 43 C, D). Látka povahy ceroidu se primárně kumuluje v lysosomech ve skupině neuronálních ceroid-lipofuscinos. K indukci ceroidu dochází u avitaminosy E z důvodu zvýšené peroxidace lipidů.
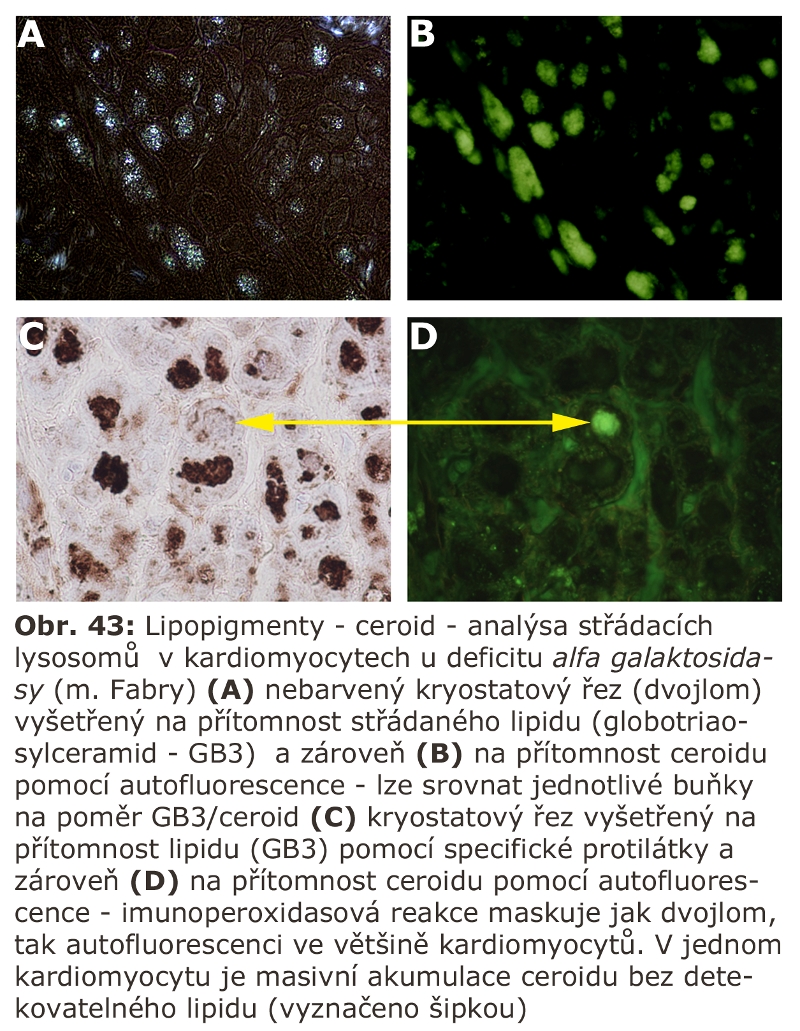
Průlomovým zjištěním bylo pozorování, že lze indukovat ceroid (v původních experimentech byl nazván lipofuscin) bakteriálním tripeptidem leupeptinem, který inhibuje lysosomální thiolové proteasy (katepsiny B, H, L) a vyvolává tak speciální obraz lysosomální proteinosy. Akumulovaný materiál je nedegradovatelný, fluoreskuje a je hydrofobní. Splňuje tedy kriteria residuálního tělíska typu ceroidu. Tyto pokusy dokázaly, že akumulace proteinu může být zodpovědná za vznik autofluorescentních residuálních tělísek. To bylo významně podpořeno zjištěním, že podstatnou součástí autofluorescentních lysosomálních tělísek ve skupině tzv. neuronálních ceroid-lipiofuscinos je podjednotka c mitochondriálního komplexu ATP syntasy (komplexu V OXFOS), ve kterém je součástí protonoforického kanálu zabudovaného do vnitřní mitochondriální membrány (viz výše - oddíl tkáňová patologie).
Jde o výrazně hydrofobní polypeptid, který vykazuje v agregovaném stavu autofluorescenci a akumuluje se z neznámých důvodů v neurolysosomech u této skupiny poruch. U některých variant neuronální ceroidlipofuscinosy jsou však dominantním střádaným proteinem v autofluorescentních lysosomálních granulích jiné hydrofobní proteiny - saposiny (aktivátory skupiny lysosomálních hydrolas (viz výše)).
Extracelulární ceroid. Shora zmíněné situace se týkají intracelulárního ceroidu, akumulovaného v lysosomech. Za některých situací dochází k deposici ceroidního materiálu extracelulárně. Zde je velmi pravděpodobný vznik z peroxidovaných lipidů.
Extracelulární ceroid vzniká v rozpadající se tukové tkáni (běžné v podkoží nebo v tzv. corpora libera v peritoneu). Histologicky jde o eosinofilní membránová deposita hyalinní povahy, vystýlající většinou rozpadové dutiny v tukové tkáni (Obr. 44 A), depozita jsou PAS positivní (Obr. 44 B). Ceroid je silně hydrofobní, barvitelný tukovou červení i v parafinových řezech (Obr. 44 C) a je autofluorescentní (Obr. 44 D). Bývá často fagocytován makrofágy, takže je pak sekundárně lokalisován intracelulárně.
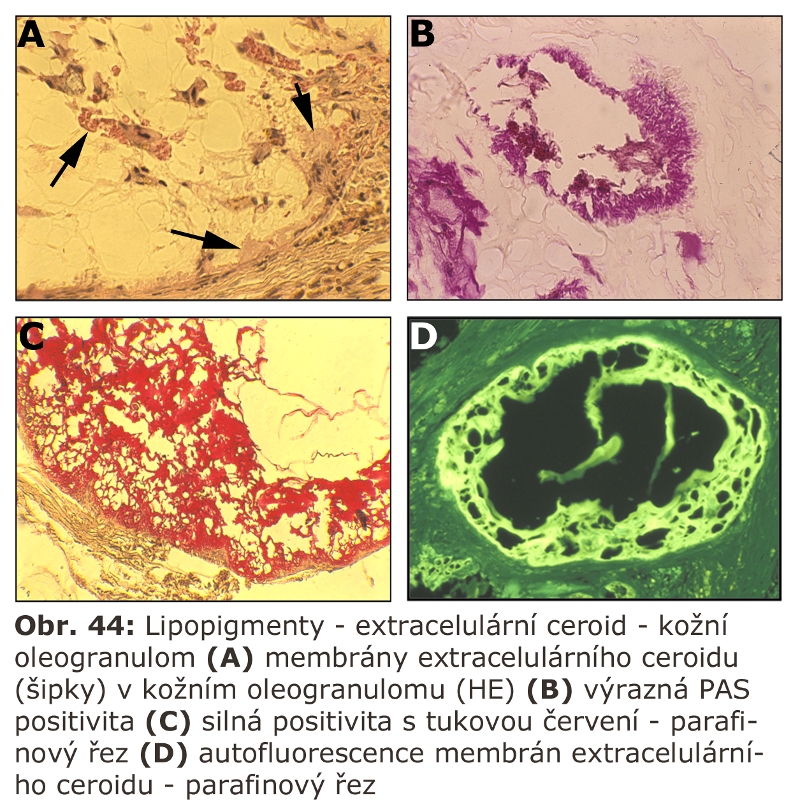
Analogicky vzniká extracelulární ceroid u Nasu-Hakolovy nemoci (lipomembranosní osteodysplasie a sklerosující leukoencefalopatie), u které, jako k jednomu z projevů, dochází k rozpadu tukové tkáně a tvorbě extracelulárního ceroidu.
Podobná deposita se vyskytují v centrech ateromových arteriálních plátů okolo nekrotických makrofágů ve formě silných undulujících membrán (Obr. 45). Prominentním znakem těchto deposit je opět výrazné barvení sudanovými barvivy v parafinových řezech a autofluorescence. Tady jsou zřejmě primárním zdrojem nekrotické lipofágy.
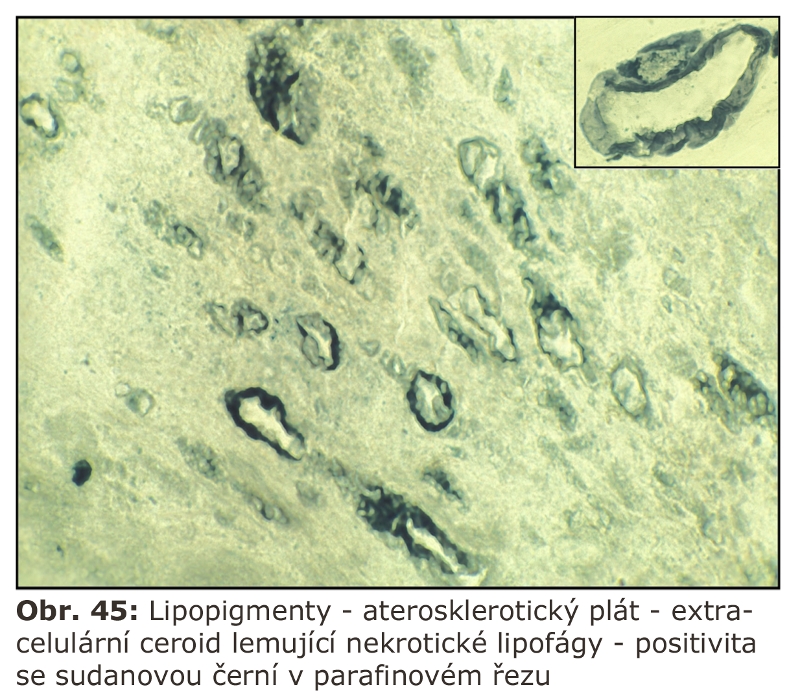
je podle současné konsensuální definice pigment, který se akumuluje v lysosomech buněk, zejména postmitotických (neurony, kosterní sval, myokard, ale i některých epiteliích) jako projev "stárnutí" což vedlo k jeho názvu "pigment ze stárnutí" (age pigment), nebo " pigment z opotřebování". Stupeň jeho akumulace je úměrný věku. Iniciální stadia deposice lze prokázat ve velmi časných obdobích života. Klasicky v potních žlázkách dětí v předškolním věku (Obr. 46). Jeho výraznou vlastností je opět, jako u ceroidu, silná autofluorescence (převážně žlutá nebo oranžová, vzácně jiná) v ultrafialovém světle a určitý (malý) stupeň hydrofobicity. Má velmi pestrou ultrastrukturu, některé z ultrastrukturálních variant jsou pro lipofuscin příznačné (Obr. 47)
Převládající hypotesou je vznik lipofuscinu z peroxidovaných lipoproteinů buněčných organel segregovaných v lysosomech autofagocytosou. Jeho zvýšená akumulace podmiňuje hnědou barvu orgánů starých lidí (tzv. hnědá atrofie), což je detailně probráno v učebnicích patologie.
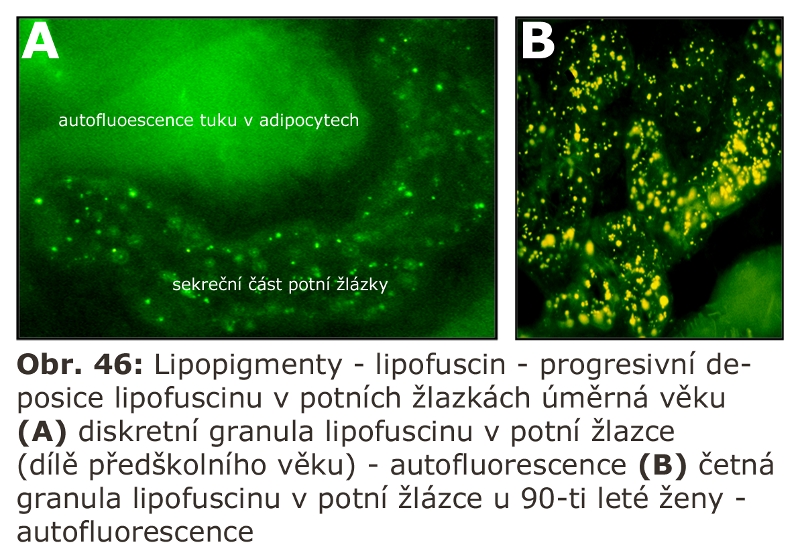
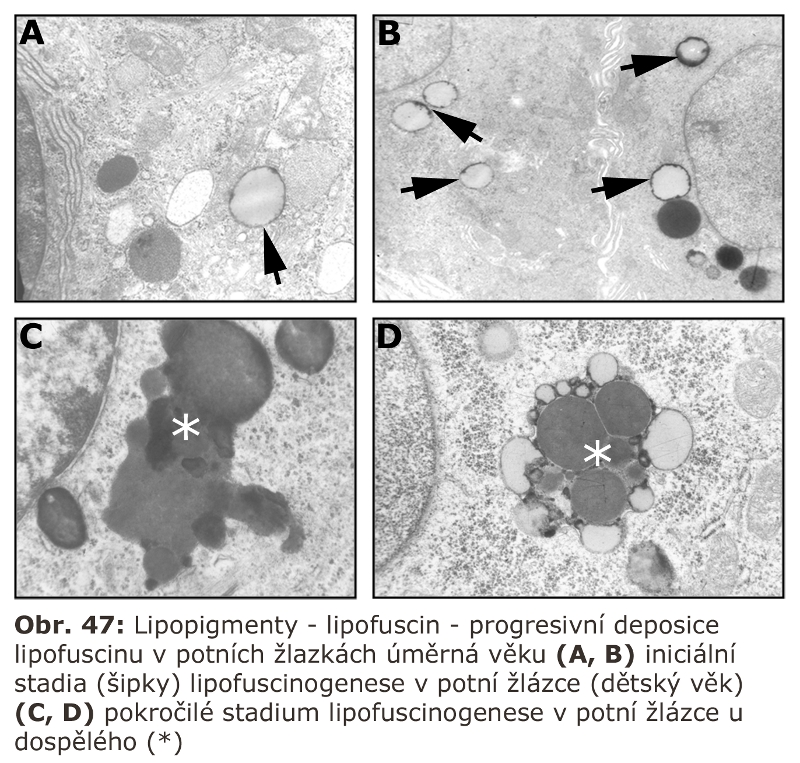
Souhrnně lze říci, že lipopigmenty jsou, jako skupina, definovány převážně fyzikálně a empiricky situacemi, za kterých se vyskytují, což je naprosto nedostatečné. Nepochybně je to skupina velmi heterogenní. Jejich přesná biochemická a molekulárně biologická klasifikace je předmětem výzkumu. Mechanismus lipoperoxidace, původně považovaný za výlučný, nelze v některých případech vyloučit, ale stále více se kumulují pozorování, že za vznik ceroidu mohou být zodpovědné hydrofobní proteiny agregované v lysosomech. Za zmínku stojí, že není doposud známa povaha fluorogenu, který zodpovídá za charakteristickou autofluorescenci.
Ve schématu 18 jsou uvedeny tinkční charakteristiky lipopigmentů (lipofuscinu i ceroidu)

Pokud jde o positivitu v barvení dle Massona (Schéma 18), jde o variantní lipopigmenty (ceroidy) známé z patologie Dubin-Johnsonova syndromu (pigmentace hepatocytů) (Obr. 48 A-D) a Heřmanského- Pudlákova syndromu a prokazatelné v makrofázích u melanosis coli (deposice v histiocytech ) (Obr. 48 E, F). Tímto připomínají redukční schopnosti melaninu. Na rozdíl od něho však tyto variantní lipopigmenty neredukují roztok argentnitrátu. Navíc jsou lysosomálně lokalisované a vykazují intensivní autofluorescenci.
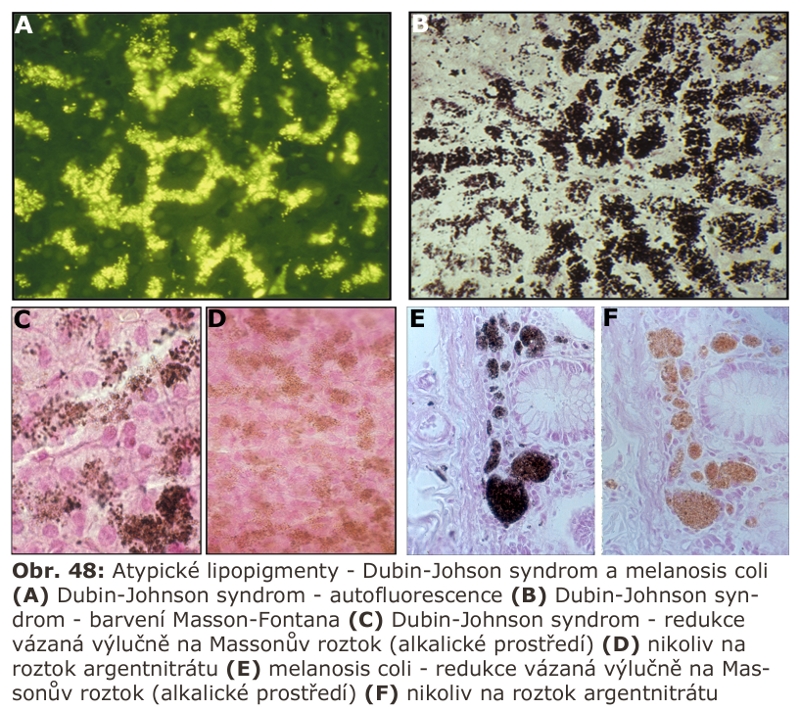
S.S. Seehafer, D.A. Pearce: You say lipofuscin, we say ceroid: defining autofluorescent storage material ( Neurobiol. Ageing 27:576-588, 2006)
S.S. Seehafer, D.A. Pearce: Spectral properties and mechanisms that underline autofluorescent accumulations in Batten disease.( BBRC 383: 237-254, 2009)
D.N. Palmer, et al.: The origin of fluorescence in the neuronal ceroid lipofuscinoses (Batten disease) and neuron cultures from affected sheep for studies of neurodegeneration. (Arch. Gerontol. Geriat. 34:343-357, 2002)
V následující části jsou probrána residuální tělíska obsahující kovy (siderosomy, kuprosomy). Vznikají za všech stavů, spojených s hypersaturací buněčného cytosolu (specifických vazných proteinů) kovem, což indukuje po delší době jejich sekvestraci v lysosomech. Mechanismus přenosu kovu z cytosolu do lysosomu není přesně znám, ale všeobecně se předpokládá, že jde o autofagocytosu.
Železo je nezbytný kov pro organismus uplatňující se v širokém spektru základních biologických funkcí, ke kterým patří např. transport kyslíku, přenos elektronů, oxidační a redukční reakce anebo syntesa DNA. Avšak chemické vlastnosti železa, které jsou tak důležité pro nesčetné biochemické reakce, mohou být pro organismy velmi nebezpečné. Při fysiologické koncentraci kyslíku a při pH 7,4 železo velmi rychle oxiduje a precipituje ve formě nerozpustných hydroxidů železa. Železo však v sobě skrývá ještě jedno závažné nebezpečí, kterým je jeho schopnost katalysovat tvorbu toxických radikálů kyslíku. Organismy proto v průběhu evoluce vyvinuly schopnost vytvářet bílkoviny, které jim umožňují získávat, přenášet a skladovat železo v rozpustných a netoxických formách. Mezi nejznámější patří transferin, transferinový receptor a feritin (bílkovina, která skladuje železo v organismu); nedávný výzkum však vedl k objevu desítek dalších proteinů, které se uplatňují v homeostase železa.
Na počátku jakékoliv diskuse o metabolismu železa je nutné zdůraznit, že lidský organismus má sklon k přetížení železem, protože neexistuje žádný fysiologický systém, který by přebytečné železo vyloučil.
V cirkulaci je železo přenášeno plasmatickým glykoproteinem transferinem, který může vázat až dva atomy Fe3+ velmi pevně, ale reversibilně. Za normálních okolností velká většina buněk organismu přijímá železo z transferinu (Fe2-transferrin má lososovou barvu, která chybí u pacientů s nedostatkem železa); výjimkou jsou duodenální enterocyty, jež přijímají anorganické železo (prostřednictvím níže zmíněného Fe2+ transportéru DMT1(viz níže) nebo hemové železo (neznámým mechanismem) a makrofágy, které katabolisují hemoglobin erytrocytů. Předání železa do buněk vyžaduje vazbu transferinu na specifické membránové receptory, která je následována endocytosou těchto komplexů. V endosomech poté dojde k aktivaci "protonové pumpy", která sníží pH v těchto organelách na 5,6. Při tomto pH ztratí transferin schopnost vázat železo, které se uvolní a je přeneseno přes endosomální membránu. Apotransferin, který při pH endosomů zůstane pevně navázán na své receptory, je přenesen zpět k buněčné membráně, kde se uvolní do plazmy exocytosou. Železo je přeneseno do nitra buňky endosomálním transportérem, který nese název DMT1 (divalent metal transporter 1). Protože substrátem pro DMT1 je pouze Fe2+, endosomy musí mít schopnost redukovat Fe3+ na Fe2+; enzymatický mechanismus této redukce byl nedávno objeven. Po přestupu přes endosomální membránu je železo distribuováno na různá místa v buňce, kde se využívá pro metabolické účely: syntesu hemoproteinů a bílkovin, které obsahují nehemové železo jako prostetickou skupinu. Velmi málo je však známo o transportu železa uvnitř buněk. Železo přijaté buňkami nad jejich metabolické potřeby je skladováno ve feritinu. Buňky obratlovců mají schopnost koordinovaně, ale divergentně, regulovat syntesu feritinu a transferinových receptorů.
Feritin, který byl poprvé purifikován českým fyziologem Vilémem Laufbergerem, je tvořen dvěma typy podjednotek, označovanými jako L ("light") a H ("heavy") ferritin. Po jejich syntese v buňkách se tyto podjednotky spontánně shluknou a vytvoří bílkovinný oktahedron obsahující 24 podjednotek, jejichž poměr se liší v různých tkáních. Holo-feritin je bílkovinná skořápka, v jejíž dutině se může uskladnit až několik tisíc atomů Fe3+ ve formě železo-hydroxyfosfátu [FeO(OH)]8[FeO(H2PO4)]; zevní průměr feritinu je přibližně 12 nm, vnitřní 8 nm. Feritinová skořápka obsahuje několik kanálů, jimiž se Fe2+ přenese do nitra bílkoviny, kde se železo oxiduje na Fe3+; k tomu je nezbytná feroxidasová aktivita H-feritinové podjednotky (inaktivace podjednotky H vede k časnému úmrtí embrya).
Železo zvyšuje tvorbu feritinu, který poté uskladňuje nadbytečný kov. Při chronickém přetížení celého organizmu železem (buď vrozeném nebo získaném) anebo při intravaskulární hemolyse či po masivním lokálním krvácení, vede robustní hromadění feritinu v buněčné cytoplasmě k indukci autofagocytosy. Feritin, po částečném odbourání své bílkovinné složky v lysosomech, se označuje jako hemosiderin. Zatímco feritin se nalézá převážně v cytoplazmě, hemosiderin je přítomen v lysosomech, které se poté označují jako siderosomy. Vyznačují se výraznou elektrondensitou i v nekontrastovaných řezech (Obr. 49). Pojem "hemosiderin" (doslova železitý pigment krevního původu) je historický název, který nevystihuje ani chemickou, ani biologickou podstatu této látky. Na rozdíl od feritinu je hemosiderin nerozpustný. Jak lze uzavřít z předchozího textu, poměr mezi železem a proteinem je výrazně vyšší u hemosiderinu než feritinu. Hromadění hemosiderinu může vést k narušení funkce nebo strukturální integrity membrán postiženého lysosomu (siderosomu) a k poškození buňky. Faktem je, že při dlouhodobém přetížení buněk železem se v siderosomech začne akumulovat paralelně lipopigment zřejmě vlivem volných radikálů indukovaných železem, a to často ve značném množství. Výsledná ultrastruktura je pak výrazně heterogenní. Pro jeho kvantitativní detekci je nutné odstranit železo dithionitem (Obr. 50). Pak lze detekovat autofluorescenci, PAS positivitu, barvení aldehydfuchsinem po preoxidaci manganistanem a různý stupeň sudanofilie (Obr. 51).
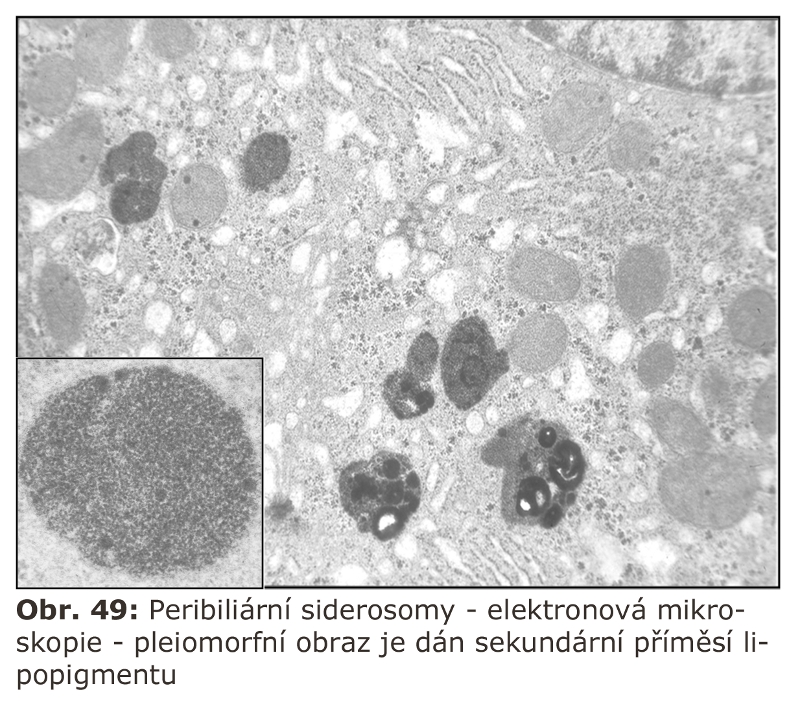
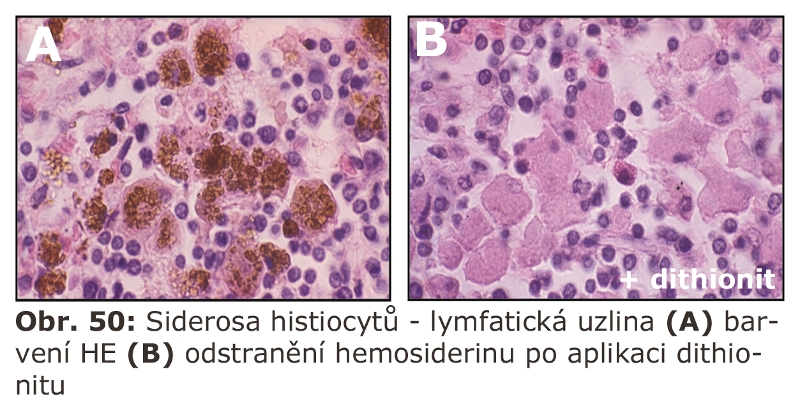
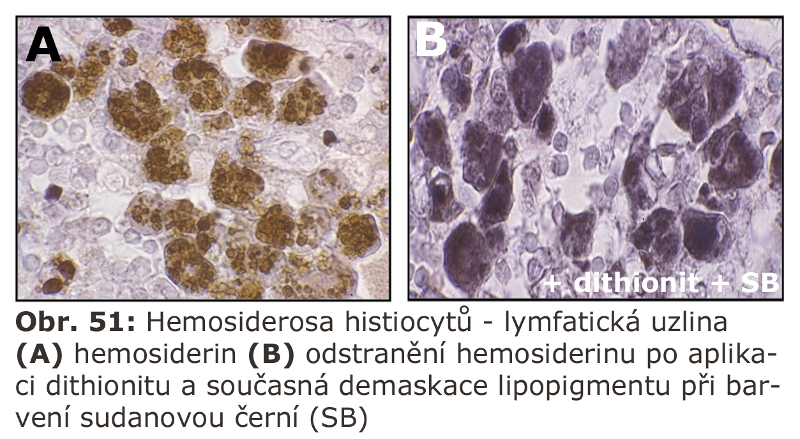
Ke zvýšení obsahu nehemového železa v buňkách a tvorbě siderosomů může dojít v důsledku (A) zvýšené dodávky nehemového železa do buněk, (B) sníženého uvolňování železa z buněk nebo (C) zvýšené dodávky hemového železa do buněk.
Ad (A) V prvním případě jde o celkové přetížení organismu železem (buď vrozené nebo získané) způsobené zvýšeným vstřebáváním železa. To vede ke zvýšené saturaci transferinu železem a často i vzniku hotovosti železa v plasmě, která není navázána na transferin. Následkem toho dojde ke zvýšení dodávky železa jak z transferinu (endocytosou), tak z netransferinové hotovosti železa (pravděpodobně prostřednictvím DMT1) do hepatocytů a jiných parenchymálních buněk a k indukci siderosomů ("parenchymal type Fe storage").
Vrozené přetížení železem se označuje jako hereditarní hemochromatosa, která může být způsobena mutacemi nejméně pěti genů. Mezi ně patří geny, které kódují HFE (jeho defekt je příčinou "klasické" hemochromatosy), hepcidin, hemojuvelin a transferinový receptor 2 (podrobnosti jsou ve speciálních textech). U všech typů hereditární hemochromatosy a mnoha typů získaného přetížení železem (viz níže) se zvyšuje vstřebávání železa.
Ad (B) Železo může opustit intaktní buňky pouze prostřednictvím membránového exportéru známého jako feroportin. Ke sníženému uvolňování železa z buněk může dojít u dvou vrozených poruch metabolismu mědi, které vedou k různým typům tkáňového přetížení železem. Jednou z nich je Wilsonova nemoc (též známá jako hepatolentikulární degenerace, viz níže) a druhou je aceruloplasminemie. Obě tyto poruchy mají blízký vztah k ceruloplasminu (lat. caeruleus = modrý), což je plasmatický α2-glykoprotein modré barvy obsahující měď. Jeho jedinou jasně definovanou fysiologickou funkcí je oxidace železa v jeho redukované formě Fe2+ na Fe3+. Feroxidasová aktivita ceruloplasminu hraje důležitou úlohu při exportu železa z buněk feroportinem. Oxidace železnatých iontů na plasmatické straně buněčné membrány poskytuje Fe3+ pro vazbu na apotransferin, čímž se zvyšuje účinnost a rychlost membránového transportu železa z buněk.
Výrazné snížení feroxidasové aktivity ceruloplasminu, ke kterému dochází u Wilsonovy nemoci (viz oddíl kuprosomy), vede k hromadění železa převážně v hepatocytech, které však není příliš výrazné; není doklad o tom, že by se železo kumulovalo v mozku. K hromadění železa v učitých buňkách mozku však dochází u druhé vrozené poruchy metabolismu mědi, aceruloplasminemie. V centrálním nervovém systému lidí a jiných savců je ceruloplasmin exprimován převážně v astrocytech, v nichž je ceruloplasmin zakotven v buněčné membráně prostřednictvím glykosylphosphatidylinositolu. Je to následkem toho, že posledních 5 C-terminálních aminokyselin, které se nacházejí v plazmatickém ceruloplasminu, je nahrazeno řetězcem 30 aminokyselin přítomných v GPI-ceruloplasminu. Vrozená aceruloplasminemie je proto doprovázena výrazným hromaděním železa v astrocytech a neurodegenerací, protože astrocyty jsou nezbytné k udržování normální funkce neuronů.
Ad (C) Ke zvýšené dodávce hemového železa do buněk může dojít buď prostřednictvím erytrocytů, hemoglobinu anebo hemu. K první situaci dochází u pacientů, kteří vyžadují léčení opakovanými transfusemi krve. Jsou to zejména pacienti s aplastickými anemiemi, myelodysplastickým syndromem a thalasemiemi, které jsou v České republice vzácné.
Zestárlé transfundované erytrocyty jsou rozpoznány makrofágy, které je internalizují procesem označovaným jako erytrofagocytosa. Fagosomy obsahující erytrocyty splynou s lysosomy za vzniku fagolysosomů. Obsažený hem se oxiduje, což vede ke snížení jeho afinity ke globinovým řetězcům. Další osud hemu není zcela objasněn, ale není pochyb o tom, že se železo uvolní z hemu enzymaticky, prostřednictvím hemoxygenasy 1 (HO1), která je přítomna v ER. Diskutují se dvě možnosti:
(i) hem se transportuje z fagolysosomů k ER, kde HO1 oxidativně rozštěpí tetrapyrolový řetězec, čímž se vytvoří biliverdin, kysličník uhelnatý a uvolní se Fe2+; (ii) HO1 je vesikulárním transportem přenesena k fagolysosomům, s nimiž splyne, a uvolní železo z hemu shora uvedeným mechanismem; Fe2+ je poté transportováno z fagolysosomů prostřednictvím DMT1 a jeho homologu, známého jako NRAMP1 (natural resistance macrophage-associated protein). Fe2+ je poté transportováno z makrofágů do plasmy prostřednictvím feroportinu. Při utlumené erytropoese (aplastická anemie, myelodysplastický syndrom) se však část železa, normálně určeného pro export, hromadí ve feritinu a později v hemosiderinu. V tomto případě se mluví o "mesenchymal" type of Fe storage - primárně makrofágy, ale později může dojít i k postižení dalších buněk.
Hemoglobin (Hb) se objevuje v plasmě při intravaskulární hemolyse, ke které dochází při některých hemolytických anemiích. Uvolněný Hb se rychle naváže na haptoglobin a poté se Hb-haptoglobinové komplexy napojí na receptory označované jako CD163 ("Hb scavenger receptors"), které jsou přítomny jen na makrofázích. Při rozsáhlejším stupni hemolysy se část hemu z Hb přenese na hemopexin; komplexy hemu-hemopexinu se poté naváží na receptory označované jako CD91 ("low density lipoprotein receptor-related protein"; "heme-hemopexin receptors"), které jsou exprimovány nejen na makrofázích, ale též na hepatocytech a fibroblastech. Po endocytose obou typů komplexů se železo uvolní z hemu prostřednictvím HO1, jak již bylo shora popsáno. Stejnými mechanismy se hemoglobin a hem zpracovávají v makrofázích a snad i jiných buňkách (např. při erytrofagocytose nádorovými krevními endoteliemi u Kaposiho sarkomu).
V této souvislosti je nutno zmínit dvě unikátní patologické situace, při nichž se nadměrné železo nenalézá ve formě siderosomů, ale hromadí se v mitochondriích. Předem je třeba zdůraznit, že u celkového přetížení železem se tento kov nehromadí v mitochondriích. Při jedné z těchto situací se železo hromadí v mitochondriích erytroblastů kostní dřeně. Nahromaděné nehemové železo lze v mitochondriích prokázat buď světelnou mikroskopií při detekci Fe3+ v kostní dřeni anebo elektronovou mikroskopií. Jelikož jsou mitochondrie přetížené železem uspořádány kolem jader v podobě prstenců, označují se tyto patologické erytroblasty jako prstenčité nebo věnečkové sideroblasty, které jsou charakteristickým symptomem u pacientů se sideroblastickými anemiemi. Patogenesa sideroblastických anemií je komplexní a, se značným zjednodušením, lze identifikovat dva základní mechanismy, které vedou ke vzniku věnečkových sideroblastů.
Všeobecně známým fenoménem jsou tzv. pseudokalcifikace, což je agregace železa v doposud ne zcela dobře specifikovaných buněčných strukturách, vzácně extracelulárně. Velmi často je tento fenomén vidět v drobných mozkových cévách (Obr. 52). K impregnaci sloučeninami obsahujícími železo dochází nejen v samotné stěně cévní, ale i v perivaskulární oblasti (velmi pravděpodobně v astrocytech), při čemž není jasno na jaké matrix a kde v buňce k deposici železa dochází. Pokud dochází k mikro hemorhagiím, hromadí se železo standardním způsobem v lysosomech makrofágů
Termín pseudovápno je odvozen od výrazné hematoxylinofilie deposit, připomínající kalcifikace. Reakce na vápno je však zcela negativní. Silně positivní je reakce na Fe3+, zcela inhibovaná dithionitem (viz níže). Afinita k hematoxylinu není změněna. Jiným, častějším příkladem z této kategorie jsou tzv. Gandy-Gamnovy uzlíky, běžné u splenomegalií. Jsou charakterisovány impregnací elastiky sloučeninami obsahujícími železo.
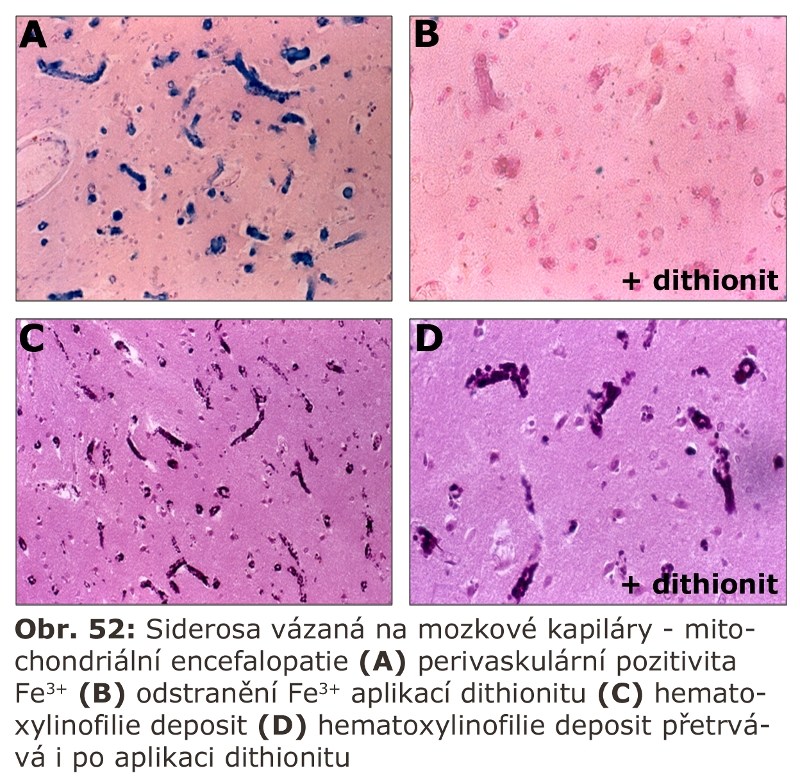
Patří sem i ferritinopatie, vzácná genetická porucha způsobená mutací genu kodujícího lehký řetězec feritinu. Dominantním projevem jsou intranukleární a cytoplasmatická tělíska složená z feritinu a dávající positivní reakci na Fe3+. Postiženy jsou neurony, glie, plexus chorioideus a celá řada dalších buněčných typů viscerálně. V běžných histologických preparátech není pigmentace železem patrná.
Pravděpodobně sem patří i skupina neuropatologických procesů, souhrnně nazývaných NBIA (neurodegeneration with brain iron accumulation), charakterisovaných, mezi jinými, akumulací železa v basálních gangliích a v subst. nigra. Zde chybí bližší informace o povaze deposit železa a jejich subcelulární lokalisaci.
Železo lze detekovat klasickým způsobem: vytvořením komplexu přímou reakcí se žlutou krevní solí (ferokyanidem) za vzniku Perlsovy modře v případě Fe3+, nebo červenou krevní solí (ferikyanidem) v případě Fe2+ (vzniká Turnbullova modř). Veškeré anorganické železo lze konvertovat na dvojmocné, vystavením tkáně sirníku, což vede k vytvoření sirníku železnatého, který je následně detekován reakcí s červenou krevní solí.
Intensitu reakce v obou klasických technikách lze zvýšit diaminobenzidinem díky redoxnímu potenciálu jak Turnbullovy, tak Perlsovy modři.
Nejcitlivější detekce železa bere v úvahu ztráty labilněji vázaného železa při imersní fixaci. Proto jako optimální je doporučeno perfundovat tkáň detekčním roztokem (žluté nebo červené krevní soli).
V detekci koordinačně vázaného hemového železa nutno použít silná oxidační činidla, klasicky koncentrovaný peroxid vodíku.
Nehemové železo (Fe3+ i Fe2+) lze spolehlivě z buněk extrahovat dithionitem, což má praktický význam, neboť umožňuje detekovat veškeré přídatné látky v siderosomu, které jsou akumulovaným železem maskovány (zejména lipopigment).
Postupy, které lze použít pro ověření kompartmentu, ve kterém se železo hromadí. V tomto smyslu se osvědčila extrakce železa nezávisle na oxidačním stupni pomocí dithionitu (viz výše). V případě lysosomální lokalisace železa lze spolehlivě prokázat lysosomální markery, zejména luminální (katepsin D), nebo známky indukce autofluorescentních residuálních lipopigmentů.
Doporučená literatura:M.W. Hentze et al.: Two to tango: regulation of mammalian iron metabolism. ( Cell 142:24-38, 2010)
A. Pietrangelo: Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. ( Gastroenterology 139:393-408, 2010)
M.D. Knutson: Iron-sensing proteins that regulate hepcidin and enteric iron absorption. ( Annu Rev Nutr.30:149-17, 2010)
M. Horvathova et al.. Molecular basis of hereditary iron homeostasis defects. (Hematology 15:96-111, 2010)
A.D. Sheftel et al.: Mitochondrial iron metabolism and sideroblastic anemia. ( Acta Haematol.122:120-133, 2009)
R. Meguro et al.. Nonheme-iron histochemistry for light and electron microscopy: a historical, theoretical and technical review. ( Arch Histol Cytol. 70:1-19. 2007)
Na basolaterálním (krevním pólu) resorbuje měď do cytosolu hepatocytu specifickým transporterem. Zde se váže na skupinu cytosolárních proteinů - metalochaperonů, bohatých na -SH skupiny, které představují přechodné depo využívané k inkorporaci mědi do metaloenzymů (cytochromoxidasa v mitochondriích, tyrosinasa v melanosomech a celá řada dalších). Další důležitý transport je směřován k specifické transportní P-ATPase 7B (ATP7B) lokalisované v oblasti trans-Golgi zóny. Odtud je měd distribuovaná k inkorporaci do apoceruloplasminu, který je sem cílen z ER-Golgi systému. Tím vzniká holoceruloplasmin, který je konstitucionální sekrecí vylučován na krevním pólu hepatocytu do krve. Při excesu mědi je transportér (ATP7B) cílen do peribiliárního vesikulárního systému na biliární pól hepatocytu (do žlučové kapiláry)
Holoceruloplasmin (ceruloplasmin s navázanou mědí) je enzym - má ferrooxidasovou funkci (oxiduje Fe2+ na Fe3+). Jeho hlavní funkcí je tedy enzymová přeměna redox stavu železa. Tím ovlivňuje i tkáňový obrat železa (viz výše), nikoliv tedy transport mědi a distribuci do periferních tkání. Tato distribuční funkce je považována za minoritní. Secernován může být i bez navázané mědi jako apoceruloplasmin, ale v této formě je poměrně rychle degradován. Holoceruloplasmin patří k tzv. proteinům akutní fáze.
Pomocí transportní P-ATPasy 7B (ATP7B) vykonává hepatocyt dvojí funkci v regulaci obratu mědi:
Zásobování tkání mědí je realisováno zejména mědí vázanou na aminokyseliny (typicky na histidin); dále existuje vazba na albumin.
Wilsonova nemoc (hepatolentikulární degenerace) se vyznačuje akumulací mědi v řadě orgánů, primárně a maximálně v játrech. Patogenetickým faktorem je blokáda sekrece mědi do žluče a její inkorporace do apoceruloplasminu, podmíněná mutací transportu P-ATPase 7B (ATP7B). Transplantaci jater lze tedy považovat za kauzální terapii. Následkem je nekontrolovaná kumulace mědi v cytosolu hepatocytů, což vede k celému spektru hepatotoxických projevů (nekrosa, steatosa, akumulace glykogenu (velmi často intranukleárně), tvorba Malloryho tělísek a další), zprostředkovaných pravděpodobně zejména indukcí volných radikálů. Nelze vyloučit, že zvýšená koncentrace mědi v cytosolu vede i k jejímu zvýšenému transportu do mitochondrií (např. cytochromoxidasa má v aktivním centru měď), což může narušit asemblaci komplexu IV. Mitochondriální dysfunkce u Wilsonovy nemoci byly posané. I ultrastrukturální abnormity mitochondrií jsou známé (Obr. 53 A). Narušené cytoplasmatické struktury (fokální cytoplasmatická degradace) jsou pak likvidovány cestou autofagocytosy, podobně jako u přetížení buňky železem (viz výše). Následkem toho dochází při dlouhodobém průběhu onemocnění k segregaci mědi do lysosomů a vytváření "residuálních" tělísek (siderosomů), na kterých participuje i lipopigment. Akumulace mědi není nikdy ale tak intensivní, aby byly kuprosomy detekovatelné v histologických preparátech, tak jak je tomu u siderosomů. Může však být detekovatelná sekundární akumulace lipopigmentu, jejíž maximum se popisuje na periferii lalůčku. Autofluorescence může být blokována akumulovanou mědí - tu lze však odstranit dithionitem, podobně jako železo (viz výše). Pro průkaz je tedy nezbytná histochemická detekce serií standardních metodik (rubeanovodíková kyselina, dimethylaminobenziliden rhodanin). Kuprosomy jsou znázornitelné i pomocí orceinu, který se váže na blíže nespecifikovaný "copper binding protein". Ultrastrukturální obraz není nikterak typický. Vzhled kuprosomu je pleiomorfní, nápadná je vysoká densita (i v nekontrastovaných preparátech) a na celkovém obraze participuje i indukovaný lipopigment (Obr. 53 B). Dosti často se v některých buňkách (hepatocytech) popisují i lysosomy s větším obsahem apolárního tuku (lipolysosomy, Obr. 53 C).
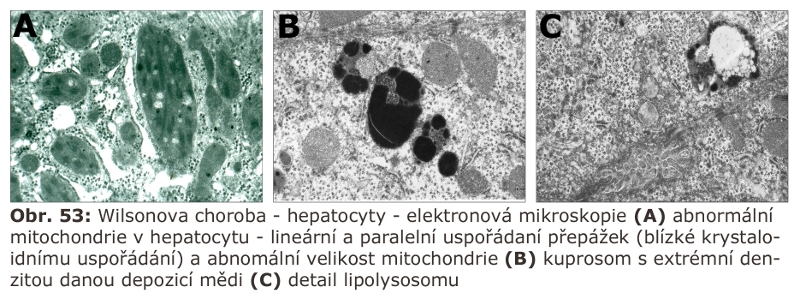
Snížená hladina ceruloplasminu v plasmě je sekundárním fenoménem u Wilsonovy nemoci. Je dána jednak poškozením hepatocytu, jednak nedostatečnou inkorporací mědi do apoceruloplasminu (chybí příslušná transportní ATPasa - ATP7B), takže se buď absolutně snižuje produkce apoceruloplasminu (jakož i ostatních plasmatických proteinů secerovaných konstitucionální sekrecí hepatocyty) nebo, pokud není produkce absolutně snížená, secernuje se jen apoceruloplasmin, který je urychleně degradován. Při výrazném nedostatku holoceruplasminu (výrazný deficit ferrooxidasové aktivity) je sekundárně ovlivněn i obrat železa. Jsou známy případy Wilsonovy nemoci, u kterých dochází k hemosiderose.
V hepatocytech může dojít ke srovnatelně intensivní akumulaci mědi i u chronických cholestas, typicky u primární biliární cirhózy. Zde je narušena primárně sekrece na žlučovém pólu. Syntesa holoceruloplasminu není narušena. Hladina sérového ceruloplasminu není tedy snížená, pokud nedojde k poškození hepatocytu cholestasou.
Zatím je nezodpověditelné, (i) jak dochází k akumulaci mědi v CNS, a jinde extrahepatálně, (ii) jaká je hladina "neceruloplasminové" mědi u Wilsonovy nemoci a (iii) na co je vázaná měď přecházející normálně do moče, zda se jedná o zmíněné aminokyseliny.
Doporučená literatura:A. Aftab et al.: Seminar, wwwthelancet.com 397, 2007
P. De Bie et al.: Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes.( J.Med.Genet. 44:673-688, 2007)
B.E. Kin et al.: Mechanisms for copper acquisition, distribution and regulation (Nature Chem. Biol. 4:176-185, 2008)
J.H. Kaplan, S. Lutsenko: Copper transport in mammalian cells:special care for a metal with special needs. (JBC 284:25461-5, 2009)
Jedním z prvých typů kontaktu bakterie s imunitně angažovanými buňkami je fagocytosa. Efektivní proces fagocytosy zahrnuje transformaci fagosomu na fagolysosom (viz výše) a optimální funkci asociovaných proteinů v membráně fagosomu. Jde o NRAMP1 (natural resistance-associated macrophage protein 1), transportér dvojmocných kovových iontů. Tento transportér se rekrutuje z buněčné membrány. Při fagocytose infekčního agens je jeho lokalisace omezena na fagosom, po jeho transformaci na fagolysosom má mizet. Je exprimován v profesionálních fagocytech: neutrofilech a makrofázích. Při jeho mutaci je tento proces nedokonalý a následkem toho dochází k nedostatečné kontrole množení fagocytovaných bakterií a k podstatně snadnější generalisaci infekce mykobacteriemi, salmonelami či leishmaniemi. Jeho funkce je vykládána jako transport dvojmocných kovových iontů z prostředí fagosomu, což snižuje vitalitu fagocytované bakterie.
NRAMP2 je ubikviterní transmembránový protein přítomný v buněčné membráně a v recyklujících endosomech. Jeho funkcí je zejména transport železa do buněk.
Do efektivního procesu fagocytosy patří i zpracování lipidních antigenů bakteriálního těla CD1 systému (viz níže)
Interference s fagosomem a jeho transformací na fagolysosom. Interakce bakterie s cílovou eukaryotní buňkou vyústí velmi často ve fagocytosu bakterie, vznik normálního fagosomu, jeho transformaci v lysosom a likvidaci pohlcené bakterie. V řadě případů (Listeria monocytogenes, ricketsie, shigelly, Bacillus subtilis) nastává degradace fagosomální membrány hydrolytickými enzymy bakterie (hemolysiny, fosfolipasami) a průnik agens do cytoplasmy. U mykobakterií je to modifikace membrány fagosomu v tom smyslu, že je znemožněna fúze s endosomálním systémem, a tedy transformace na fagolysosom. Mykobakterie tak žijí v persistujícím fagosomu v podstatě neovlivněny. Podobně je tomu v případě Toxoplasma gondii.
Porucha digesce ve fagocytární vakuole může být však způsobena i nedostatečnou funkcí T-lymfocytů (nedostatkem lymfokinů, stimulujících makrofágy). Tato skutečnost by měla být brána v úvahu u všech infekcí s nedokonalou digescí bakterií.
Speciální příčinou je dědičně podmíněná absence fenomenu respiračního vzplanutí v granulocytech v procesu fagocytosy (granulomatosa chlapců) jako následek molekulárního defektu membránově lokalisované NADPH-oxidasy. V lysosomech histiocytů se tato porucha velmi často manifestuje tvorbou značného množství ceroidu.
Při interakci cílové eukaryotní buňky s viry a vzniku infekce hraje receptorově zprostředkovaná endocytosa zásadní úlohu, neboť dle současných znalostí tímto mechanismem proniká většina virů do buňky. Prvním předpokladem vstupu viru do buňky je jeho přichycení na buněčný receptor, který často podmiňuje tropismus viru. Následuje vlastní průnik virionu do buňky, který spočívá ve virem indukované fúzi virového obalu a endosomální membrány, která bývá podmíněna změnou konformace fuzogenního proteinu a je buď závislá na kyselém pH (pH-dependentní) nebo na pH nezávislá (pH-independentní).
Část obalených virů vstupuje do buňky přímo pomocí fúze virového obalu s cytoplasmatickou membránou následované uvolněním virové nukleokapsidy do cytoplasmy (paramyxoviry, herpesviry, HIV-1) ev. pomocí makropinocytosy (poxviry). Většina ostatních virů využívá ke vstupu do buňky klatrin-dependentní či independentní endocytosu. Po fúzi virového obalu s membránou endosomu následuje uvolnění a přestup nukleokapsidy do cytoplasmy, odtud případně translokace do jádra. U obalených virů dochází k fúzi virového obalu s membránou endosomu na různých úrovních endosomálně lysosomálního systému v závislosti na pH. Neobalené viry vstupují do buňky rovněž endocytosou a z endosomu se dostávají pomocí acidifkace endosomu, teplotně-závislou změnou konformace a tvorbou póru nebo pomocí proteolýsy v lysosomu.
a) presentace proteinových antigenů pomocí systému MHC
b) presentace lipidních antigenů pomocí systému CD1
Podle klasické definice jsou antigeny endogenního původu (tj. degradační produkty buněčných či virových proteinů syntetisovaných pomocí translačního aparátu buňky) presentovány pomocí MHC I, zatímco exogenní antigeny procesované ELS jsou presentovány pomocí MHC II
Presentace pomocí MHC I (každá buňka v těle) se děje exocytosou transportních vesikul Golgiho aparátu na povrch buňky. Tyto váčky obsahují transmembránově zakotvené MHC I, na které jsou navázány peptidy, určené k presentaci. Tyto peptidy jsou připraveny z proteinů v cytosolu, a to naštěpením v tzv. imunoproteasomu (tento je odlišný od standardního proteasomu a produkuje peptidy vhodnější k navázání na MHC I komplex). Tyto "na míru šité" imunogenní peptidy jsou transportovány speciálními membránovými ABC transportéry TAP (transporter associated with antigen processing) do ER, kde jsou navázány na molekuly MHC I systému a transportními váčky dopraveny na buněčnou membránu. Tento systém není navázán na ELS. Antigeny presentované pomocí MHC I jsou rozpoznávány CD8+ T-lymfocyty (detaily jsou ve specialisovaných textech)
V případě presentace pomocí MHC II (makrofágy, dendritické buňky, B lymfocyty) dojde k fúzi váčků ELS (obsahujícími částečně proteolyticky zpracovaný proteinový antigen) s transportními váčky z trans-Golgi zóny, obsahujícími transmembránově zakotvený komplex MHC II molekul. Fúzí těchto dvou typů váčků vznikne hybridní MHC II kompartment, ve kterém dojde k navázání antigenních peptidů na MHC II komplex (je považován za součást LRO systému viz níže). Tyto hybridní váčky jsou určeny k exocytose s cílem umístit komplex MHC II a antigenního peptidu na povrch presentující buňky. Tyto antigeny exogenního původu (tj. fagocytované či endocytované) presentované pomocí MHC II jsou rozpoznávány CD4+ T-lymfocyty.
Existují však důkazy proto, že mnohé z těchto exogenních antigenů jsou presentovány pomocí MHC I a slouží k presentaci CD8+ T-lymfocytům. Tento způsob se nazývá cross-presentace a dochází k němu in vivo v dendritických buňkách. In vitro jsou cross-presentace schopny i makrofágy a B-lymfocyty. Avšak mechanismy, kterými se exogenní antigeny dostanou na MHC I, jsou stále nedostatečně známé. V současnosti se rozeznávají 2 způsoby, jak se exogenní antigen dostane z endosomu či fagosomu do transportních vesikul a na povrch buňky, kde je presentován pomocí MHC I (i) Cytosolární dráha (TAP a proteasom-dependentní). Exogenní antigen či antigen intracelulárních bakterií a parasitů množících se v některém z kompartmentů ELS dostane do cytosolu pomocí transportních kanálů v ELS membránách antigen presentujících buněk. V cytosolu je ubikvitinován a parciálně procesován v imunoproteasomu a dále transportován do ER (viz výše); (ii) existuje předpoklad, že může dojít přímo k fúzi ELS váčků, obsahujících částečně proteolyticky zpracovaný protein, přímo s MHC I transportními váčky tak, jak je tomu v případě presentace pomocí MHC II (viz výše), takže cytosolární kompartment s proteasomem se cross-presentace neúčastní (tzv. vakuolární dráha, TAP a proteasom independentní).
Vzhledem k tomu, že zejména viry často intereferují s presentací svých peptidů na povrchu napadené buňky klasickým MHC I či s presentací pomocí antigen presentujících buněk (APC), je cross-priming důležitý pro rozpoznání a likvidaci virové infekce pomocí CD8+ T-lymfocytů. Ve srovnání s viry je stimulace CD8+ T-lymfocytů antigeny intracelulárních bakterií či parasitů výrazně slabší, což zřejmě souvisí s jejich vlastním proteosyntetickým aparátem, který je oddělený od ER.
Ad b) presentace lipidních antigenů pomocí systému CD1Tento systém představuje druhý systém molekul presentujících antigen. Antigenem jsou v tomto případě lipidy, a to buď endogenní nebo exogenní. Tato presentace je součástí vrozené imunity, ale významě ovlivňuje i adaptivní imunitní odpověď. Lipidní antigen je pomocí CD1 systému presentován T-lymfocytům (non-NK T-lymfocytům a i NK T-lymfocytům). Podle současných představ hraje aktivace T-buněk antigenními lipidy důležitou roli v detekci a eliminaci různých patogenů, přičemž je významná téměř okamžitá aktivace a produkce Th-polarisujících cytokinů.
CD1 molekuly jsou transmembránové proteiny (typ I integrálního membránového proteinu), resp. glykoproteiny. Jsou exprimovány v raftových mikrodoménách na povrchu antigen presentujících buněk (B lymfocyty, makrofágy, dendritické buňky). Zde jsou v komplexu s β2-mikroglobulinem. U člověka existuje pět typů CD1 molekul, kódovaných pěti geny (CD1a-e), v každém z nich existuje několik isoforem.
Dynamika tohoto systému není doposud zcela jasná. Molekuly CD1 jsou v ER a Golgiho aparátu postranslačně upravovány glykosylací, za dohledu chaperonů asociovány s β2-mikroglobulinem a exocytosou dopraveny na buněčnou membránu. Odtud jsou dopravovány endocytosou do různých etáží ELS, kde se konjugují s lipidními molekulami a jsou zpětně exocytosou dopraveny na povrch buňky. Ve skupině antigenních lipidů zastupují významnou roli sfingolipidy a jejich deriváty. V současném pohledu nastává vazba CD1 komplexu na exogenní lipid v endosomálním systému, tedy až po endocytose nebo fagocytose lipidní molekuly (připouští se do jisté míry i přímá vazba antigenního lipidu na CD1 molekuly na povrchu buňky, nebo již v endoplasmatickém retikulu, pokud jde o vlastní lipidy). V procesu exposice lipidu hrají roli saposiny, deriváty prosaposinu a NPC proteiny (viz výše). Interakce T-lymfocytu s CD1 systémem indukuje proces, který vede k usmrcení cílové buňky nebo k indukci cytokinové odpovědi.
Tento proces hraje významnou roli v imunitě proti bakteriálním infekcím, kdy jsou antigenní lipidy z fagocytovaných bakterií (exogenní lipidy) takto exponovány a indukují reakci T-lymfocytů. Klasickým příkladem jsou lipidy M. tuberculosis (mykolová kyselina a řada jejích derivátů) exponované na povrchu makrofágů a vedoucí k jejich destrukci T-lymfocyty. Jde ale i o antigenní lipidy (zejména glycerofosfolipidy) pylu, zodpovědné za alergii na pyl. Mezi potenciálně antigenní tělu vlastní lipidy patří sulfatid a isoGB3 (isoglobotriaosylceramid). Nejvíce studovaným antigenním CD1 vázaným lipidem je α-galaktosylceramid, původem z mořských hub.
Pro další podrobnosti o problematice molekulární biologie CD1 molekul odkazujeme na příslušnou literaturu
Doporučená literatura:G. De Libero, L. Mori: How the immune system detects lipid antigens ( Prog. in Lipid Res. 49:120-127, 2010)
N.R.. Cohen et al.: Antigen presentation by CD1: lipids, T cells, and NKT cells in microbial immunity. ( Advances in Immunology 102, 2009)
M. Salio et al.:Recent advances in processing and presentation of CD1 bound lipid antigens. ( Current Opin. Immunol. 22:81-88, 2010)
Tento systém cytoplasmatických sekrečních granul sdílí mnohé rysy s klasickým ELS. V jeho membránách jsou membránové komponenty lysosomálních membrán (LAMP proteiny, naopak MPR chybí), obsahem granul jsou v mnoha případech i lysosomální enzymy samotné, přestože degradace biokonjugátů nebyla nikdy v systému LRO přesvědčivě prokázána. Všechna LRO granula obsahují celou řadu biologicky aktivních látek určených k regulované sekreci. Mezi nejlépe definovaná granula tohoto systému patří (Obr. 54)
Řadí se sem i Weibel-Palladeho granula endotelií, spermatický akrosom, lysosomy napojené na presentaci antigenu (MHC II systém), sekreční granula juxtaglomerulárního aparátu, blíže nespecifikovaná granula makrofágů a buňky proximálních kanálků ledvin (bez specifikace granul), a několik dalších buněčných struktur.
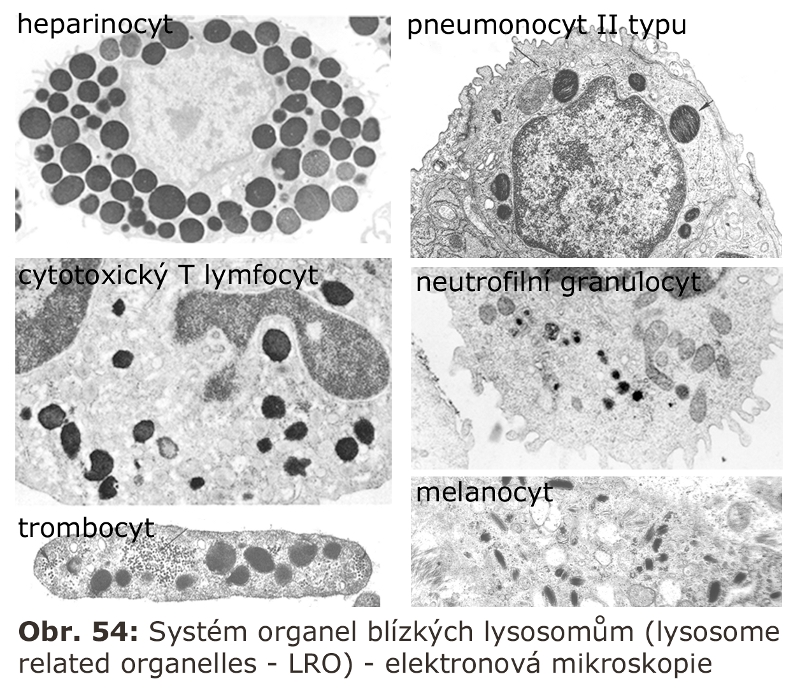
Biogenese celého systému má společný buněčně biologický základ s ELS. Membránové komponenty LRO granul jsou dopravovány jako transmembránové kargo pomocí specifických adaptorových proteinů. Jejich multipodjednotkový systém, deficitní u Heřmanského-Pudlákova syndromu, se nazývá BLOC (biogenesis of lysosomes-related organel complex). Transportní váčky pocházejí buď přímo z trans-Golgi zony nebo do LRO putují přes časný endosom, kde se stýká ELS s LRO. O selektivním importu luminálních proteinů, které by mělo zajistit specifitu cílení, není nic podstatného známo.
Exocytosa LRO je regulována celou řadou cytosolických faktorů (rab proteiny, myosiny a další)
Je tedy zřejmé, že tzv. lysosomální kompartment není jediný z oddílů buňky, ve kterém se vyskytují kyselé hydrolasy a komponenty lysosomálních membrán. Jinak řečeno přítomnost lysosomálních enzymů nebo membránových lysosomálních komponent v určité struktuře nemusí nutně znamenat, že jde o strukturální součást degradačního ELS. Blízký vztah granul LRO a ELS naznačuje i existenci společných regulačních cest, které mohou a jsou velmi často společně postiženy v rámci známých genetických poruch LRO.
Genetické poruchy LROlze obecně definovat jako poruchy biogenese LRO nebo poruchy regulace LRO exocytosy. Hlavní klinické následky jsou uvedeny pouze v souhrnném pohledu (Schéma 20):

Strukturálně se porucha manifestuje přítomností abnormálně velkých granul LRO. Jde především o abnormálně velká azurofilní granula v neutrofilních leukocytech. Dále jsou přítomna abnormální sekreční granula v krevních destičkách, heparinocytech, pneumocytech II typu. Abnormální jsou i sekreční granula hlavních buněk ve sliznici žaludku a exokrinního pankreatu. Porucha se manifestuje i na úrovni melanocytárních melanosomů, z nichž některé mohou taktéž být abnormálně zvětšené. Paralelně jsou postiženy i lysosomy některých dalších buněk (neuronů, hepatocytů, histiocytů nebo kanálků ledvinných). Akumuluje se v nich ceroid, jehož biochemická podstata není známa (viz residuální tělíska). Na poruchu lze po stránce funkční nazírat jako na selhání mechanismů kontrolujících nejen velikost těchto granul, ale i jejich regulovanou exocytosu.
Diagnostická jsou abnormálně velká primární granula neutrofilů (Obr. 55)
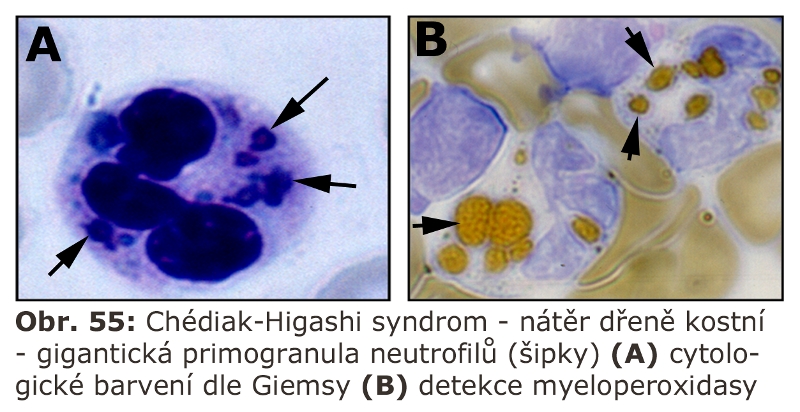
Syndrom je geneticky heterogenní - u myši je známo přes 15 genů, které mohou být isolovaně mutované, a tak vést k rozvoji HPS (Obr. 56), u člověka takových genů bylo zatím identifikováno osm. Jde o mutace v systému BLOC (viz shora). Mikroskopicky je patrná výrazná ceroidosa makrofágů a absence melaninu v epidermis. V destičkách chybí densní granula. Sekreční granula pneumonocytů jsou abnormálně veliká. Popisována je také granulomatosní kolitida, nefropatie a plicní fibrosa.
Existuje několik málo dalších jednotek (Griscelliho syndrom, Elejaldeho syndrom), které jsou podrobně rozvedeny ve specificky zaměřených publikacích.
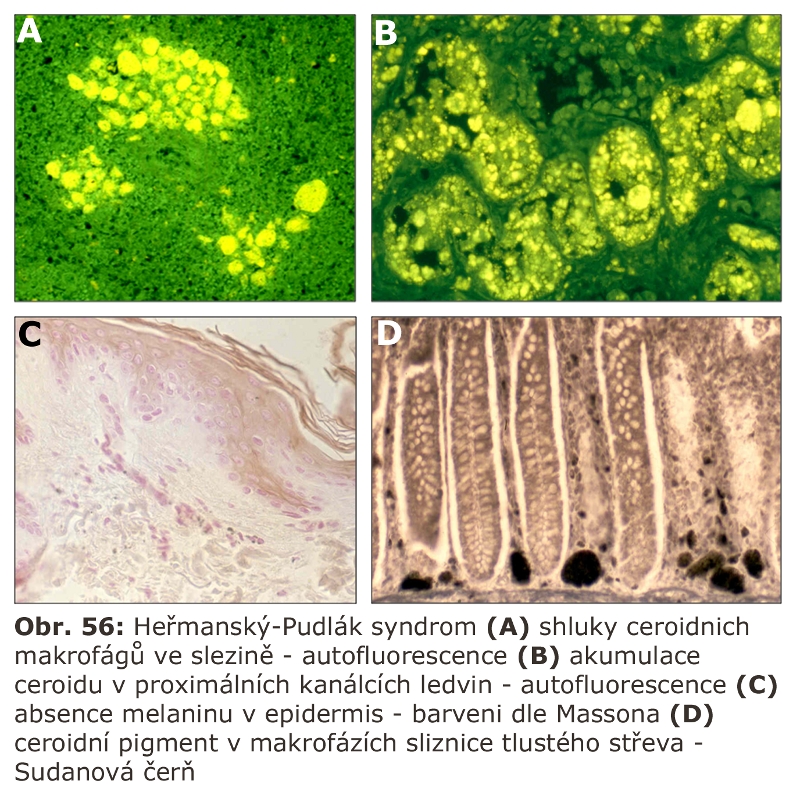
M. Huizing et al.: Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. ( Annu Rev.Genomics Hum.Genet. 9:359-386, 2008)
Existuje oprávněný předpoklad, že s vývojem základního biomedicinského výzkumu budou objeveny další jednotky, jejichž podstatou bude dysfunkce lysosomálního systému, která se nebude manifestovat lysosomálním střádání, ale dysfunkcí jiného typu.
Jako příklady lze uvést:Cystatiny a jejich deficit. Cystatiny jsou inhibitory lysosomálních proteás. Jejich deficit zvyšuje jejich aktivitu. Např. deficit cystatinu B vede z progresivní neurologické poruše známé jako Lundborg Unverrichtova epilepsie (Baltic myoclonus). V mozku jsou popisovány pouze degenerativní změny. Další definovanou jednotkou je deficit tranlysosomálního transportéru pro vitamin B12 (hydroxykobalaminu)
| příjmení | jméno | narozen | zemřel | poznámka |
| Batten | Frederick Eustace | 1865 | 1918 | anglický neurolog a pediatr |
| Bielschowsky | Max | 1869 | 1940 | německý neuropatolog, emigroval z Německa do Velké Británie |
| Dorfman | Ronald F. | žijící | americký patolog | |
| Elejalde | Rafael | kolumbijsko-americký genetik | ||
| Fabry | Johannes | 1860 | 1930 | německý dermatolog |
| Farber | Sidney | 1903 | 1973 | americký pediatr |
| Gamna | Carlo | 1866 | 1950 | italský lékař |
| Gandy | Charles | 1872 | 1943 | |
| Gaucher | Philippe Charles Ernest | 1854 | 1918 | francouzský dermatovenrolog, histolog a patolog |
| Griscelli | Claude | 1936 | francouzský pediatr | |
| Hakola | Panu | finský lékař | ||
| Heřmanský | František | 1916 | 1980 | finský lékař |
| Higashi | Otokata | japonský pediatr | ||
| Hunter | Charles A. | 1873 | 1955 | skotský lékař |
| Hurlerová | Gertruda | 1889 | 1965 | německá pediatryně |
| Chediak | Alexander Moises | 1903 | kubánský lékař a serolog | |
| Janský | Jan | 1873 | 1921 | český neurolog a psychiatr, profesor |
| Krabbe | Knud Haraldsen | 1885 | 1905 | dánský neurolog |
| Kufs | Hugo Friedrich | 1871 | 1955 | německý neuropatolog |
| Lamy | Maurice Emile Joseph | 1895 | 1975 | francouzský lékař a genetik |
| Maroteaux | Pierre | 1926 | francouzský pediatr | |
| Morquio | Luis | 1867 | 1935 | uruguayský pediatr |
| Nasu | T. | japonský neuropatolog | ||
| Niemann | Albert | 1880 | 1921 | německý pediatr, profesor, vedoucí dětské nemocnice Berlin-Hallensee |
| Pick | Ludwig | 1868 | 1944 | německý patolog, profesor, vedoucí oddělení patologické anatomie Friedrichshain-Berlin, pro židovský původ perzekuován nacisty, zemřel v ghettu Terezín |
| Pompe | Johannes Cassanius | 1901 | 1945 | holandský patolog, popraven nacisty za účast v holandském odbojovém hnutí |
| Pudlák | Pavel | český hematolog, ÚHKT Praha | ||
| Rosai | Juan | žijící | americko-argentinský patolog | |
| Sachs | Bernard | 1858 | 1905 | americký neurolog a psychiatr |
| Sandhoff | Konrad | 1939 | německý biochemik, profesor, Univerzita v Bonnu | |
| Sanfilippo | Sylvester | 1926 | americký pediatr | |
| Santavuori | Pirkko | žijící | finský neurolog, profesor, University of Helsinky | |
| Scheie | Harold Glendon | 1909 | 1990 | americký oftalmolog |
| Schindler | Detlev | žijící | německý pediatr, biochemik, genetik, Universitat Wurzburg | |
| Sly | William S. | žijící | americký biochemik a molekulární biolog | |
| Spielmeyer | Walter | 1879 | 1935 | německý neurolog a psychiatr |
| Tay | Warren | 1843 | 1927 | anglický oftalmolog a chirurg |
| Vogt | Heinrich | 1875 | 1936 | německý neurolog |
| Wolman | Moshe | 1914 | izraelský neuropatolog, narozen ve Varšavě |