Molekulární patologie lipidů v nejobecnějším smyslu slova zahrnuje veškeré poruchy metabolismu lipidů. Velká většina těchto stavů však nemá přímý strukturální odraz, ale může se manifestovat sekundárními poruchami buněčných funkcí a celým řetězcem dalších změn. Příkladem je celá řada stavů, při nichž došlo ke kvalitativním a kvantitativním (tj. včetně patologicky snížené syntesy) změnám spektra lipidů (např. glykolipidů, mastných kyselin polárních lipidů, steroidů, apod.) existujících membránových struktur nebo jejich mikrodomén (raftů)
Obsah této kapitoly je zaměřen na klasické téma patologie zaměřené na stavy vedoucí k akumulaci lipidů do takové míry, že je detekovatelná optickým mikroskopem, v řadě případů i prostým okem. Nepočítáme-li specielní problematiku lipidos (viz níže), je celá široká problematika steatos centrována na triacylglyceroly a estery cholesterolu. Úvodem nástin normální situace u jednotlivých lipidů, kterých se poruchy týkají a základních buněčných procesů, ve kterých jsou tyto dvě základní skupiny lipidů zapojeny.
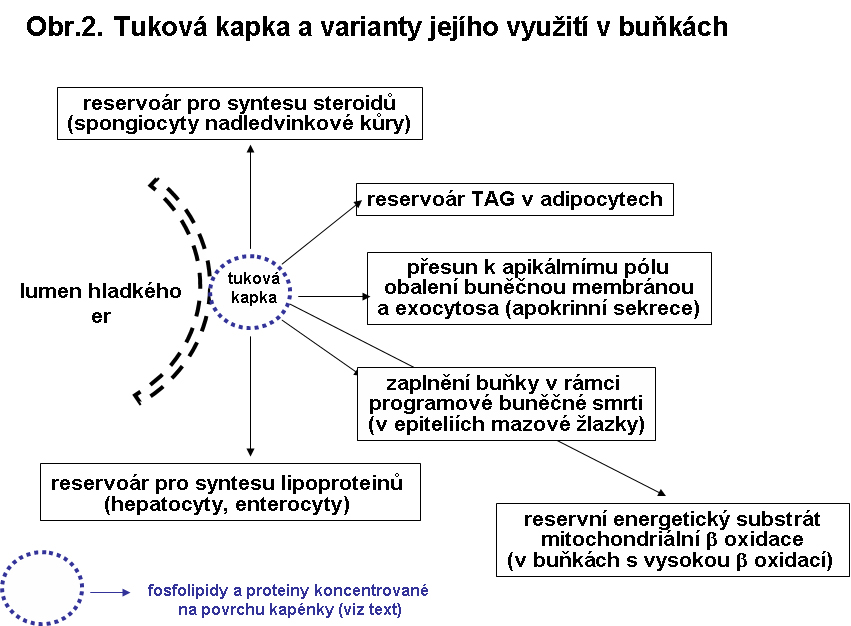
Triacylglyceroly (TAG), jakožto nositelé mastných kyselin, jsou ve velkém kvantu v buňkách bílé a hnědé tukové tkáně, kde slouží jako energetická reserva. Kapky TAG mohou být přítomny fysiologicky v malém množství prakticky ve všech buňkách, zejména v buňkách s intensivní β oxidací mastných kyselin jako reservní substrát. V hepatocytech může být dosti výrazná reserva TAG deportovaná z tukové tkáně. V játrech normálního dospělého člověka se odhaduje obsah TAG v rozmezí 6.7 gr - 60.7 gr. Množství může významně stoupat za hladovění, což je způsobeno mobilizací volných mastných kyselin z bílé tukové tkáně. Reserva TAG slouží jednak k syntese lipoproteinů v hepatocytech pro export (viz níže).Velké depo triacylglycerolu je i perisinusoidních Itových buňkách jater sloužících jako reservoár vitaminu A.
Kapky TAG jsou přítomny i ve žlázkách secernujících tuky. V případě mazových žlázek jde o progresivní multilokulární akumulaci TAG v cytosolu, končící zánikem buňky (holokrinní typ sekrece). Dle některých autorů se lipid iniciálně hromadí v endoplasmatickém retikulu. V případě laktující mléčné žlázy jsou kapénky TAG dirigovány cytosolem k apikálnímu pólu buňky, kde se před uvolněním obalí buněčnou membránou (apokrinní typ sekrece).
Estery cholesterolu (ChE) jsou přítomny ve formě cytoplasmatických kapének v buňkách specialisovaných na syntesu steroidních hormonů. V cytoplasmě jsou lokalizovány v cytosolu. Jsou zde jako reservoár cholesterolu, který je výchozím stavebním kamenem všech steroidů. Typicky je tomu ve spongiocytech kory nadledvin, do kterých je cholesterol dopravován LDL receptorem zprostředkovanou endocytosou.
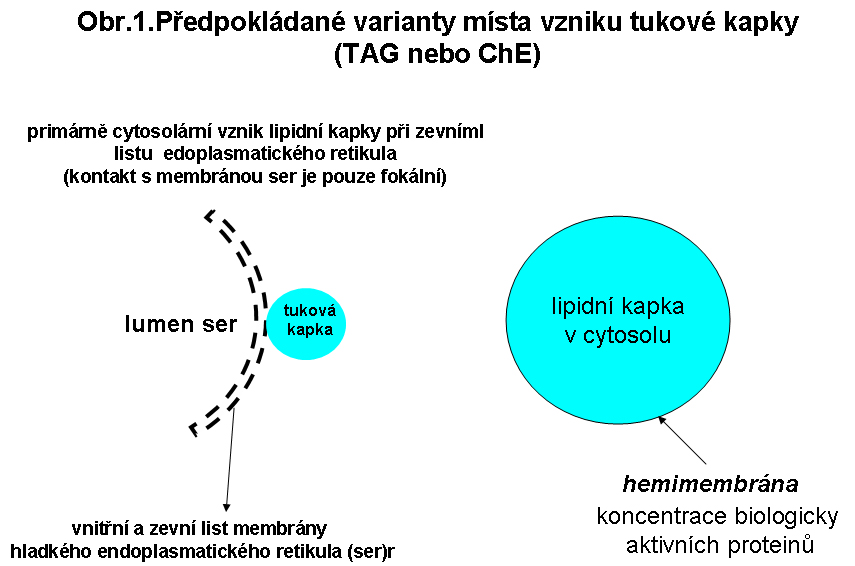
Tukové kapky - jejich biologieLipidní kapénky TAG ChE jsou lokalizovány v cytosolu. K esterifikaci glycerolu a cholesterolu esterifikaci dochází enzymy nacházející se v membránách endoplasmatického retikula (hladké endoplasmatické retikulum), které je hlavním kompartmentem řídícím obrat lipidních esterů tukové kapky, která se nachází v cytosolu. V membráně endoplasmatického retikula jsou enzymy acyltransferázy, přenášející mastnou kyselinu z Acyl-koenzymu A na cholesterol (Acyl koenzym A: cholesterol acyltransferasa, ACAT ) nebo na glycerol, resp. diacylglycerol (diacylglycerolacyltransferasa, DAGT). Existuje též názor, že tyto tukové kapky vznikají přímo v membráně hladkého ER (mezi oběma listy - cytosolárním a luminálním) a jsou de facto intramebranosní. Během prvé fáze se předpokládá intramebranosní akumulace, která se může šířit laterálně mezi listy, pokud není nastartován vlastní proces pučení do cytosolu.pod vlivem jednoho z proteinů (ADRP viz níže), který vznikající kapku obaluje.
Na povrchu cytosolárních kapének (obr.1), tedy na rozhraní lipidní estery - cytosol, jsou koncentrovány fosfolipidy, zejména lecitin, včetně lysolecitinu, volný cholesterol a řada biologicky aktivních proteinů. Pro tuto vrstvu je používán termín hemi-membrána lipidní kapky. Výčet proteinů koncentrovaných na povrchu lipidní kapky je dlouhý. Za zmínku stojí skupina PAT zahrnující perilipin, ADRP (adipofilin - adipocyte differentiation related protein) a TIP47. Některé z nich hrají významnou roli v regulaci degradace lipidních esterů v kapce. Prokázáno to bylo v případě dvou geneticky podmíněných stavů charakterizovaných progresivní akumulací TAG v cytosolu (viz níže)
perilipin je protein exprimovaný výhradně v adipocytech a ve steroidogenních buňkách; chrání estery lipidní kapky před neregulovanou hydrolysou; zprostředkuje vazbu lipázy (adipocyte triglyceride lipase - ATAGL, známou též jako PNPLA2 a pod dalšími zkratkami) na tukovou kapku. Pro funkci této lipázy je nezbytná asociace s proteinem CG1-58, který se váže na perilipin.Těmito vazbami je zaručena vazba lipázy na tukovou kapku (přenos z cytosolu) a její aktivita. Deficit CG1-58 byl prokázán u Chanarin-Dorfmannova syndromu (viz níže). Deficit ATAGL (lipázy) má za následek syndrom blízký Chanarin-Dorfmanovu syndromu - neutral lipid storage disease (střádání neutrálních lipidů - míněny jsou pouze TAG). Kapének esterů cholesterolu se tyto poruchy netýkají. Funkce ADRP a TIP47 nejsou doposud dostatečně známé.
Speciální zapojení cytosolární tukové kapky lze sledovat v buňkách secernujících lipoproteiny (viz níže), v buňkách s apokrinní sekrecí (průnik cytosolem k apikálnímu pólu buňky, její obalení buněčnou membránou a odškrcení) a buňkách mazových žlazek (zaplnění celé buňky, která následně programově zanikne a vyprazdňuje obsah do vlasového foliklu). Způsoby různých zapojení tukové kapky jsou shrnuty v následujícím schematu
Lipidy plicního surfaktantu. Speciálním případem je sekrece lipoproteinů plicního surfaktantu. Zde jsou fosfolipidy (zejména lecitin) syntetizovány na zevním listu membrány endoplasmatického retikula, transportovány do jeho lumen a dále za přidání dalších komponent do Golgiho aparátu a ve formě membránou ohraničených granul konstitucionálním typem sekrece předávány do alveolů. Tato granula bohatá na lipidy jsou de facto variantou lysosomálních granul (systém LRO - lysosome related organels - viz lysosomální systém)
Sekrece sfingolipidů v granulární vrstvě epidermis. V buňkách stratum gramulosum jsou sfingolipidy (glukosylceramid) a koncentrované do sekrečních mikrogranulí (Odlandova tělíska), která jsou exocytována do mezibuněčnéno prostoru. Tam je secernována i glukocerebrosidasa. Tím je nastartován prvý krok k lipidní (hydrofobní) bariery epidermis v rohové vrstvě.
Mechanismy vedoucí k nadměrné akumulaci hlavních druhů lipidů triacylglycerolů a esterů cholesterolu1. s poruchou beta oxidace mastných kyselin v mitochondriích. Pokud nejsou mastné kyseliny v mitochondriích oxidovány musí být kovalentně vázány (na glycerol, viz níže poruchy beta oxidace). Akumulovaný TAG je tedy v těchto případech nepřímým projevem poruchy spalování mastných kyselin. Nutno zmínit, že buňka, která je vystavena přísunu mastných kyselin z krve nebo která syntetisuje nadměrné množství mastných kyselin, kdy se tedy hromadí mastné kyseliny, které nejsou v daném okamžiku utilisovatelné, jsou deponovány ve formě TAG (v neesterifikované formě ohrožuje buněčné struktury detergentním účinkem)
2. s poruchou syntesy (asemblace) lipoproteinů v endoplasmatickým retikulu, určených pro export do celého organismu. Jde prakticky výlučně o hepatocyty a enterocyty (viz též níže). Tato porucha postihuje všechny molekuly inkorporované do molekuly lipoproteinů. Akumulovány jsou však pouze TAG - jde tedy o primární poruchu utilisace TAG na rozdíl od poruch beta oxidace, kdy jsou akumulované TAG výrazem prevence toxického účinku volných mastných kyselin
Pokud jde o cholesterol, je jeho akumulace za řady stavů dána neschopností buňky akumulovaný cholesterol degradovat. Pokud není inkorporován do novotvořených membrán, nebo odstraněn z buňky
V následujícím textu budou zmíněny hlavní doposud známé mechanismy, které představují základní cesty metabolismu lipidů, při jejichž poruše dochází k patologické akumulaci tuku v buňce. V rámci různých nosologických jednotek se mohou tyto mechanismy kombinovat. Selhání uvedených mechanismů může být i absolutní (jejich poruchou) nebo relativní (nadměrnou zátěží).
Pokud nebereme úvahu poruchy syntesy lipidů (er-Golgi, peroxisomy), pak se na těchto poruchách zúčastní tyto buněčné oddíly: endoplasmatické retikulum, mitochondrie a lysosomy.
I. Syntesa lipoproteinů a jejich transport - fysiologie a patologieEndoplasmatické retikulum (syntesa a sekrece lipoproteinů - speciálních transportních forem lipidů). Podle dnešních znalostí jsou lipoproteiny syntetizovány v dvojím typu buněk.
Proces syntesy lipoproteinu probíhá v hladkém endoplasmatickém retikulu (ER). Kvantitativně nejvýraznější součástí jsou TAG, syntetizované v membráně endoplasmatického retikula a deponované v membráně, případně v cytosolárních kapkách, obsahující TAG (viz shora). U secernovaných lipoproteinů je tedy důležitá kooperace mezi luminem ER a cytosolem přes membránu ER. Prvým krokem je translokace ApoB do lumen ER (v oblasti RER) a jeho postupná lipidisace (v oblasti SER), ve které dominuje transport TAG přes membránu ER z cytosolární lipidní kapky (viz shora) nebo z jejího mezi membránového prostoru, katalyzovaný MTP (mikrosomální TAG transferový protein); přibírán je cholesterol, fosfolipidy. Finální VLDL partikule je pak mikrovesikulrním systémem transportována do Golgiho aparátu , kde podléhá dalším posttranslačním modifikacím. Z terminální části Golgiho aparátu (transgolgi zóna) jsou pak lipoproteinové agregáty (velikost 30 - 80 nm) transportovány ve formě sekrečních vesikul na basolaterální membránu enterocytů a hepatocytu (vaskulární - basolaterální pól obou buněčných typů) a secernovány do krve (lymfy). Vzhledem k značné velikosti partikulí chylomikronů jsou finální sekreční váčky v enterocytech mnohem více zřetelné. Proces sekrece lipoproteinů patří do kategorie sekrece konstitutivní. Takto vzniklé lipoproteiny, představující mikroemulsi koloidních kapének, slouží jako vehikulum pro transport lipidů do periferních tkání.
Poznámky. MTT je heterodimer tvořený protein disulfid isomerasou (PDI) a další podjednotkou o 97 kD Směr procesu v enterocytech jde od pólu apikálního (resorpce mastných kyselin z lumen) na basolaterální pól, kde je exocytosa; naproti tomu směr procesu v hepatocytech na krevním basolaterálním pólu začíná i končí Hepatocyty jsou spolu s adipocyty považovány za jediné dvě buňky schopné vytvářet reservní depo tuku. Na tukovou tkáň (bílou tukovou tkáň) je vhodné pohlížet jako na "trvalé centrální depo", na hepatocyty jako na "přechodné depo distribuční", kde jsou připravovány transportní (lipoproteinové) formy tuku. Existují pozorování svědčící pro možnost sekrece lipoproteinů kardiocyty (byl v nich prokázán transkript jak pro Apo, tak pro MTT)
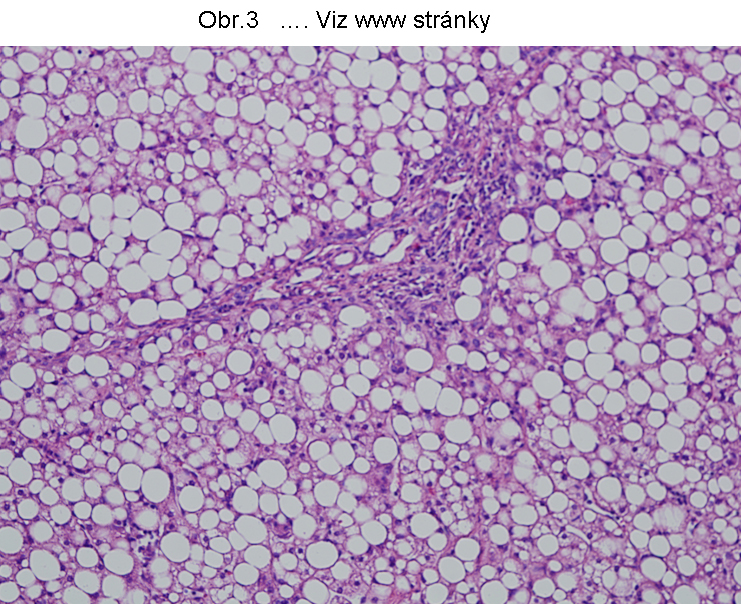
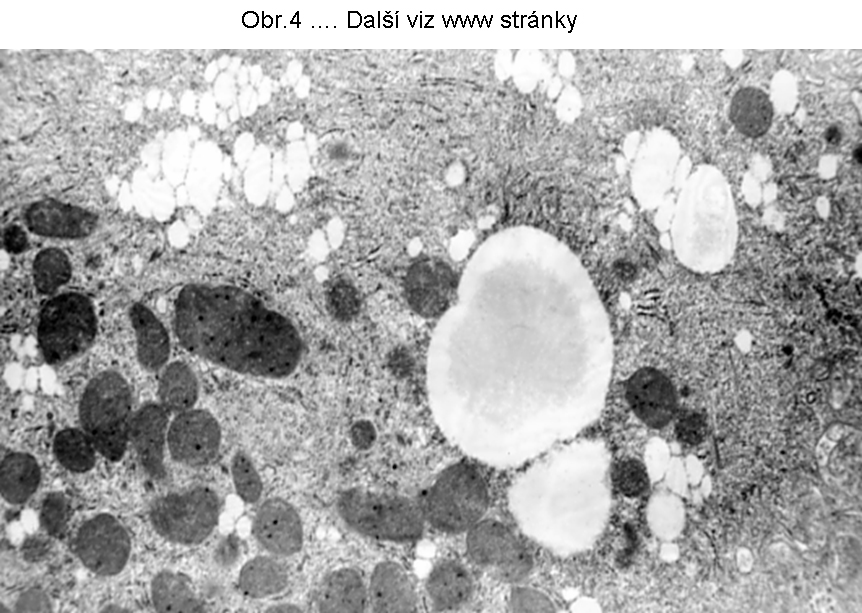
PatologieTato steatosa je v současném pohledu omezena na hepatocyty a enterocyty (buňky syntetisující lipoproteiny), které jsou v současné době jedinými známými elementy, schopnými syntetisovat a secernovat lipoproteiny krevní plasmy (viz poznámku níže). Tukové kapky se skládají z TAG a nacházejí se v cytosolu (Obr.3, 4, 5).
Největší frekvence výskytu této steatosy je v hepatocytech (obr.3). K narušení vysoce koordinovaného procesu lipoproteinové syntesy v játrech může dojít za nejrůznějších okolností , např. nedostatkem lipotropních faktorů, porušením endoplasmatického retikula a tím syntesy apoproteinu B (nebo MTP viz shora a níže), působením toxických látek, případně infekce. Genetické poruchy jsou zmíněny níže. Rozvoj této poruchy se manifestuje steatosou hepatocytu (enterocytu), při čemž k akumulaci tuku dochází v cytosolu (obr.4), tedy ve formě tukových kapének, jejichž propojení s membránou ER je prokázáno. Akumulace lipidu v lumen endoplasmatického retikula nebyla u tohoto stavu přesvědčivě prokázána.
Poznámka. V tomto smyslu patří tento proces do kategorie poruch sekrece, definovaný poruchou asemblace komplexní molekuly v ER, srovnatelný s poruchami typu "endoplasmic reticulum storage disease, viz poruchy sekrece).Jako "storage" lze definovat cytosolární depo nevyužitého lipidu
Genetické poruchy procesu sekrece lipoprotenů1. Defektní syntesa (asemblae) lipoproteinů v endoplasmatickým retikulu - retence TAG v cytosolu
V současnosti jsou známé poruchy způsobené mutací v genu MTP (viz shora). Důsledkem je defektní asemblace VLDL v hepatocytech a chylomikronů v enterocytech. V obou buněčných typech se rozvíjí steatosa. Lipidní kapky jsou deponovány v cytosolu. Apo B je zvýšen na úrovni transkriptu, ale protein je degradován v ER nebo v proteasomu (jako důsledek "neutilisace" v ER). Immunocytochemicky je ale v příslušných buňkách prokazatelný.
Sekundárně se rozvíjejí následky malabsorpce, avitaminosy E (myopatie, kardiomyopatie, neuropatie, retinopatie) a důsledky změny složení membránových lipidů erytrocytů (ostnité erytrocyty - akantocytosa)
Dále jde o autosomálně recesivní poruchu danou mutací v samotném apoB genu. Patogeneticky a fenotypově jde o poruchu blízkou deficitu MTP proteinu (viz výše). Steatosa postihuje též hepatocyty a enterocyty. Tukové kapky jsou lokalizovány v cytosolu (Obr.5). Následkem je snížená sekrece lipoproteinu.
Porucha vesikulárního transportu lipoproteinu po jeho asemblaci v endoplasmatickém retikulu - retence lipidu v transportních vesiklech
2. Chylomicron retention disease je samostatná porucha omezená na enterocyty. Podstatou je porucha vesikulárního transportu asemblovaných chylomikronů transportními vesikly z oblasti er do Golgiho aparátu. Lipid se akumuluje v transportních vesiklech, což dosahuje rozměru rozpoznatelného v optickém mikroskopu (Obr.5). Mutace postihuje rab protein Sar1b kodovaný SAR1B genem (dříve zvaným SARA2). Tento protein se spoluúčastní v komplexu COP II proteinů na regulaci vesikulárního transportu z er do Golgiho aparátu v enterocytech. V plasmě chybí chylomikrony a apoB 48. Produkce hepatálních lipoproteinů (VLDL apo B 100) probíhá nerušeně a jsou v séru prokazatelné. Nedávno bylo poukázáno na možnou manifestaci deficitu i v dalších orgánech (kosterním svalu, myokardu), vzhledem k tomu, že COP II komplex se zúčastňuje vesikulárního přenosu i mimo enterocyty.
Také je možno předpokládat, že budou identifikovány další varianty poruchy vesikulárního transportu chylomikronů z úrovně er-golgi (místo asemblace lipoproteinu) až k basolaterálnímu pólu enterocytu. Tím by spadala tato skupina poruch do široké kategorie poruch exocytosy (konstitucionální sekrece) a mohla by být v různém rozsahu spojena s poruchou exocytosy v řadě dalších buněčných typů.
Obr. 5. Schema základních mechanismů steatos daných poruchami syntesy a sekrece lipoproteinu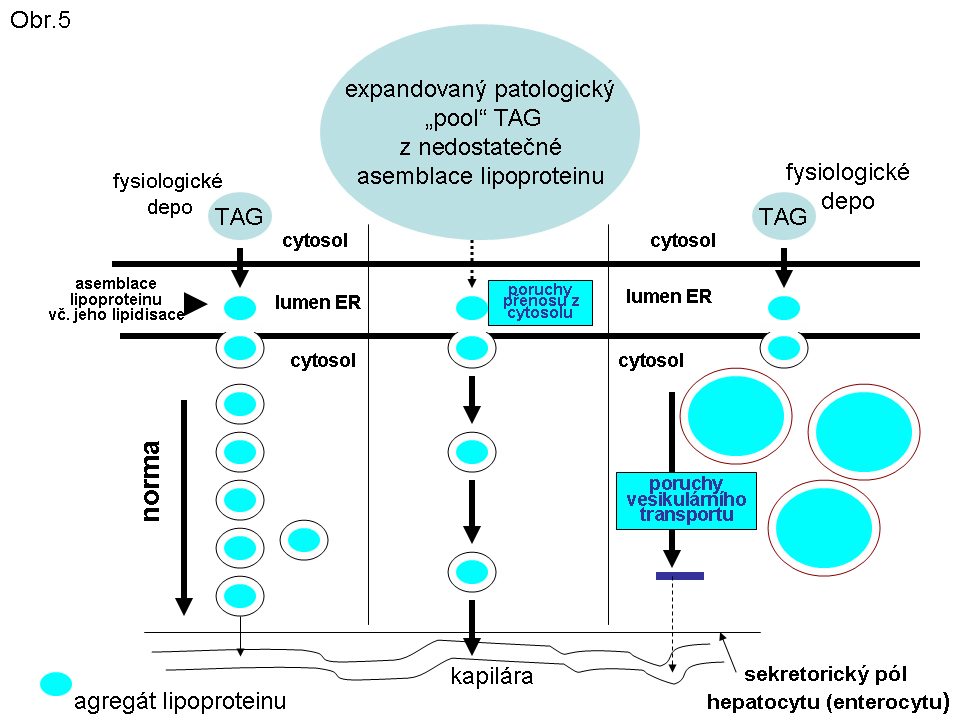
Přechod MK s dlouhým řetězcem (C18 -C12) přes buněčnou membránu je podpořen komplexem volně asociovaných proteinů, které přenášejí MK z albuminu, na který jsou vázány v krvi, do buňky. V cytosolu jsou MK vázány po průchodu buněčnou membránou na tkáňově specifické transportní proteiny a jimi distribuovány. Transport MK s dlouhým řetězcem do mitochondrie je umožněn kaskádou enzymových reakcí. Nejdříve je mastná kyselina vázána na koenzym A. Následně je acyl MK přenesen na karnitin karnitinpalmitoyl transferasou I (CPT I). Tento přenos nevyžaduje aktivní transport vzhledem k poměrně volnému propojení vnitřního mezimembránového prostoru mitochondrie s cytosolem. CPT I se vyskytuje ve třech isoformách: jaterní, svalové a mozkové. Jaterní isoforma se liší tím, že je velmi citlivá na inhibici malonyl-CoA, což umožňuje v játrech dle energetických potřeb MK odbourávat nebo syntetizovat. Po této prvé konjugaci následuje aktivní přenos přes vnitřní membránu (translokasou) a pak znovu konjugace s koenzymem A (karnitinpalmitoyl transferasou II - CPT II) a následná β-oxidace (viz obr.6). Mastné kyseliny se středně dlouhým řetězcem (C12-C6) a s krátkým řetězcem (C4-C6) přechází do buňky a do mitochondriální matrix prostou difúzí. V mitochondriích probíhá proces tzv. β- oxidace (BOX) mastných kyselin. Mastné kyseliny s dlouhým řetězcem, středně dlouhým řetězcem a krátkým řetězcem jsou oxidovány odlišnými sadami enzymů, kodovanými z odlišných genů. Na zkrácení MK o dva uhlíky se podílejí vždy 4 procesy:1.dehydrogenace, 2. hydratace vzniklé dvojné vazby, 3.další dehydrogenace vzniklého hydroxyacylu a 4. thiolása (jde de facto o Acyl-CoA acyltransferasu, vnášející CoA do zbytku a odstraňující uvolněný acetyl vázaný na CoA). Poslední tři procesy jsou u mastných kyselin s dlouhým řetězcem katalysované jediným, tzv trifunkčním multienzymem, což je heterooktamer, složený ze 4 alfa a 4 beta podjednotek (u zkrácených řetězců jde o tři individuální enzymy).
Obr.6. Schéma procesů, podílejících se na transportu mastné kyseliny do β oxidační spirály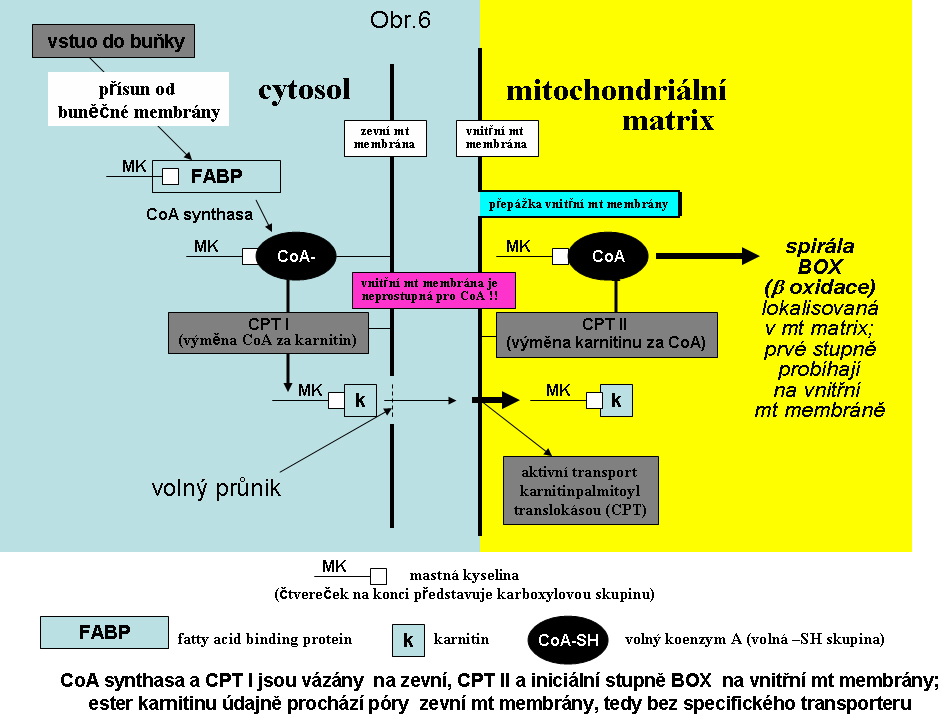
V průběhu β-oxidace se uvolňuje sada acetyl-CoA, které přecházejí do Krebsova cyklu, dále z prvých fází β-oxidace (flavinové dehydrogenasy) jsou přenášeny redukované flavinové koenzymy (FADH2) přes ETF (electron transfer flavoprotein) další dehydrogenasou (ETF- ubichinon oxidoreduktasou) do respiračního řetězce v místě Ubichinonu. Redukované NADH z hydroxyacyldehydrogenas třetí fáze β-oxidačního cyklu jsou přenášeny na komplex I respiračního řetězce. Z posledních dvouhlíkatých zbytků (acetyl-CoA) jsou syntetisovány ketolátky - aceton, acetoacetát a β-hydroxybutyrát (proces restringovaný na játra). β-hydroxybutyrát je předáván do cirkulace je využíván celou řadou tkání, zejména neurony. V nich je β-hydroxybutyrát redukován na acetoacetát, kondensován s CoA a na spálen v Krebsově cyklu.
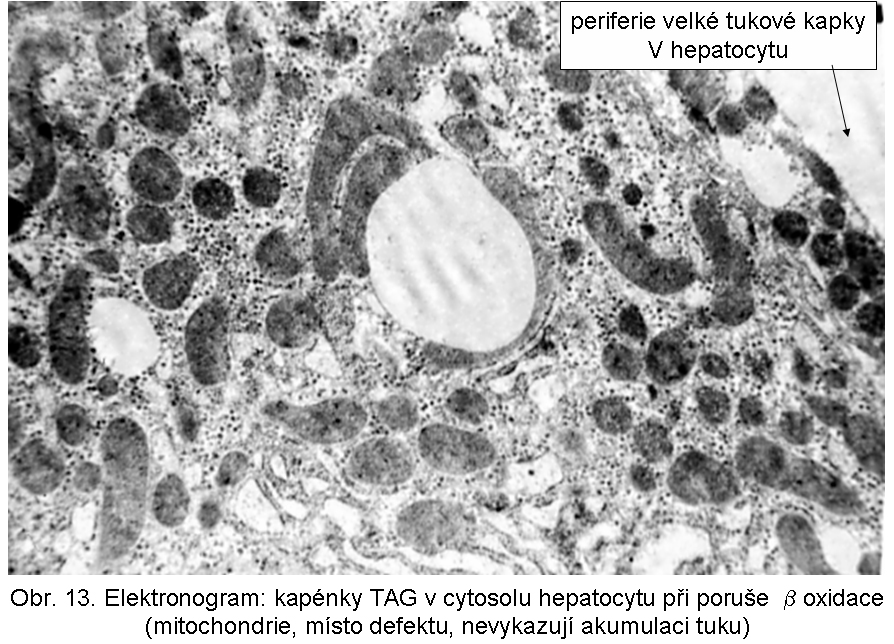
Poruchy beta oxidace (BOX; v anglosaské lit FAO - fatty acid oxidation)Porucha se projevuje zvláště intensivně v buňkách s vysokou energetickou spotřebou, pro které jsou vodíkem bohaté řetězce mastných kyselin energeticky nejvhodnějším substrátem. Jde zejména o srdeční a kosterní sval, ledviny a hepatocyty. Akumulované, neutilisované MK s normální délkou řetězce (C16-C18) jsou v cytoplasmě okamžitě reesterifikovány na TAG a ve formě kapének deponovány v cytosolu buňky. Paralelně mohou být kondensovány s karnitinem a vylučovány jako acylkarnitiny. Kapénky TAG mají tendenci být vázány k mitochondriím, což je zvláště dobře patrné v buňkách s topicky segregovanými mitochondriemi (např. basální žíhání v buňkách proximálně stočených kanálků.
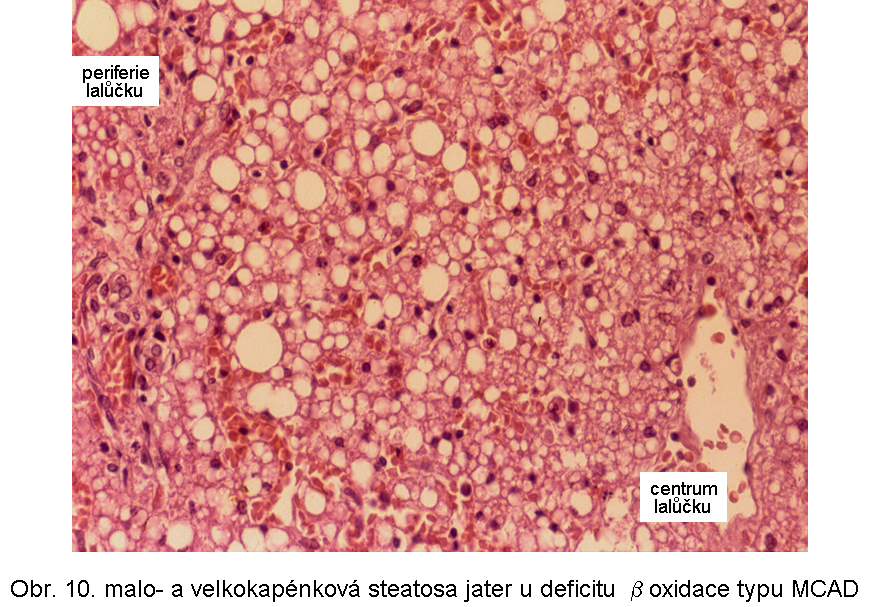
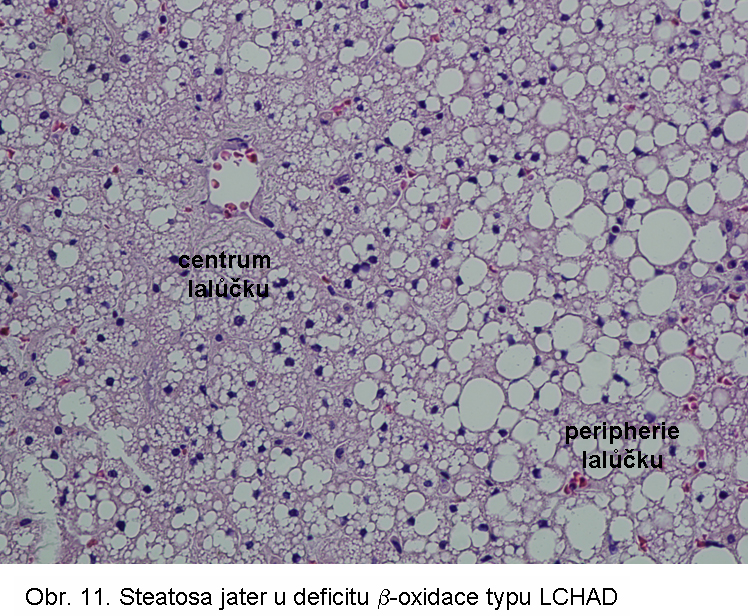
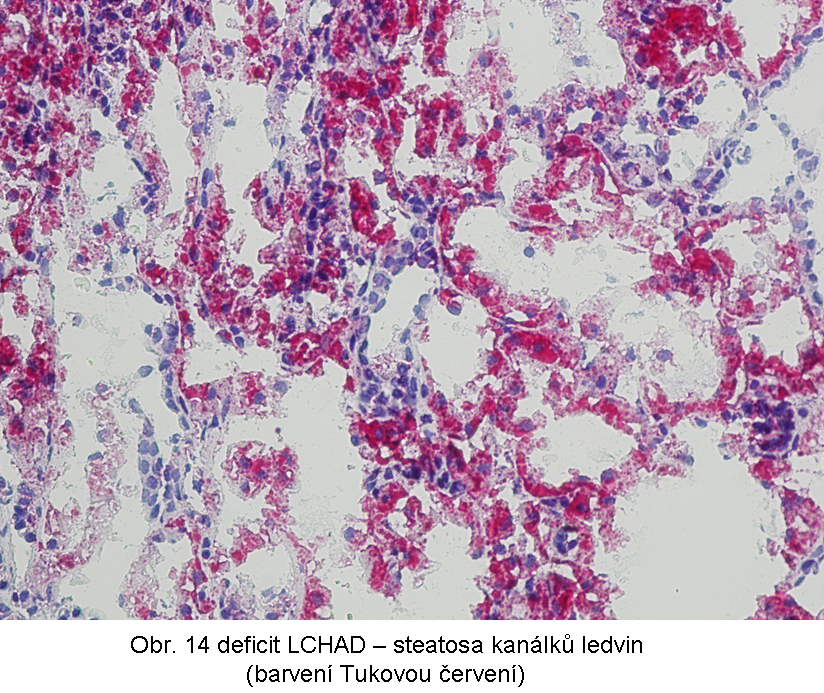
Porucha je způsobena následujícími základními mechanismy:Primární dědičné poruchy mitochondriální β-oxidace se manifestují ve všech typech poruch (s výjimkou deplece karnitin). Mezi nejčastější patří deficit dehydrogenasy acyl-CoA se středně dlouhých řetězcem (MCAD) a dehydrogenasy 3-hydroxyacyl-CoA s dlouhým řetězcem (LCHAD).
Obr. 7. Přehled definovaných genetických poruch| Deficitní protein | Zkratka | Rok popsání | MIM |
| Karnitinový přenašeč | CTD | 1988 | 212.140 |
| Přenašeč mastných kyselin přes cytoplasmatickou membránu | FATP | 1998 | - |
| Karnitinpalmitoyltransferasa I + | CPT I | 1988 | 255.120 |
| Karnitin:acylkarnitintranslokasa | CACT | 1992 | 212.138 |
| Karnitin:palmitoyltransferasa II | CPT II | 1973 | 255.110 |
| Dehydrogenasa.acyl-CoA s krátkým řetězcem | SCAD | 1987 | 201.470 |
| Dehydrogenasy acyl-CoA se středně dlouhým řetězcem | MCAD | 1982 | 201.450 |
| Dehydrogenasy acyl-CoA s dlouhým řetězcem | VLCAD | 1993 | 201.475 |
| Dehydrogenasy acyl-CoA s dlouhým řetězcem ACAD9 | ACAD9 | 2007 | 611.126 |
| Dehydrogenasa 3-hydroxyacyl CoA s krátkým řetězcem | SCHAD | 1991 | 601.609 |
| Dehydrogenasa 3-hydroxyacyl CoA s dlouhým řetězcem | LCHAD | 1989 | 600.890 |
| Mitochondriální trifunkční protein++ | MTP | 1992 | 143.450 |
| Dehydrogenasa elektrony přenášejícího flavaproteinu | MAD ETFQO | 1985 | 231.675 |
| α podjednotka elektrony přenášejícího flavaproteinu | MAD α-ETF | 1985 | 231.680 |
| β podjednotka elektrony přenášejícího flavaproteinu | MAD β-ETF | 1990 | 130.410 |
| 2,4-dienoyl-CoA-reduktáza | DER | 1990 | 222.745 |
| Thiolázáza 3-ketoacyl-CoA se středním řetězcem | MCKAT | 1997 | 602.199 |
| 3-hydroxy-3-metylglutaryl-CoA-syntáza | HMG-CoA S | 1997 | 142.940 |
| 3-hydroxy-3-metylglutaryl-CoA-ligáza | HMG-CoA L | 1976 | 246.450 |
+ existují 3 isoenzymy CPT I jaterní (CPT IA), svalová (CPT IB) a mozková (CPT IC)
++ jde o oktamer složený ze 4 podjednotek alfa a 4 podjednotek beta
Nutno dodat, že deficit beta oxidace může být sekundární v případě primárních genetických poruch oxidativní fosforylace. Získané poruchy při hypoxii, ischemii, jsou popsány na konci této části
Histologicky a v mnoha případech i makroskopicky viditelná steatosa je výrazem celkově sekundárně inhibované beta oxidace, způsobené nejspíše vyvázáním koenzymu A neoxidovatelnými mastnými kyselinami. Z toho resultuje nespecifické hromadění běžných mastných kyselin (C16, C18) a jejich reesterifikace na TAG, přestože primárně nejsou enzymy β oxidace na úrovni jejich délky řetězce postiženy.
Při poruchách β-oxidace mastných kyselin dochází k depleci poolu volného koenzymu A, protože je vázán na nezpracované acyly mastných kyselin. Koeznym A je proto uvolňován přenosem acylů MK na karnitin nebo glycin za tvorby acylkarnitinů a acylglycinů působením různých transferáz v mitochondriální matrix. Vzniklé acylkarnitiny se hromadí v buňkách a zejména ty s dlouhým řetězcem mohou působit toxicky. Acylglyciny a zejména acylkarnritiny s navázanou mastnou kyselinou s krátkým nebo středně dlouhým řetězcem jsou vyloučeny do moče. V buňce jsou acylkarnitiny s mastnou kyselinou s dlouhým řetězcem také transportovány do peroxisomů. V peroxisomech jsou MK s dlouhým řetězcem jsou odbourávány jinou sadou enzymů β-oxidací až na kyselinu oktanovou za vzniku acetyl-CoA, peroxidu vodíku, ale bez tvorby ATP. Větvené MK jsou v peroxisomech odbourávány α-oxidací. MK se středním řetězcem, jejichž zdrojem mohou být také acylkarnitiny z mitochondrie, mohou podléhat mikrosomální ω-oxidaci za vzniku dikarboxylových kyselin. Ty jsou pak β-oxidací v peroxisomech odbourávány na dikarboxylové kyseliny s krátkým řetězcem a vyloučeny do moče (viz obr. 8).
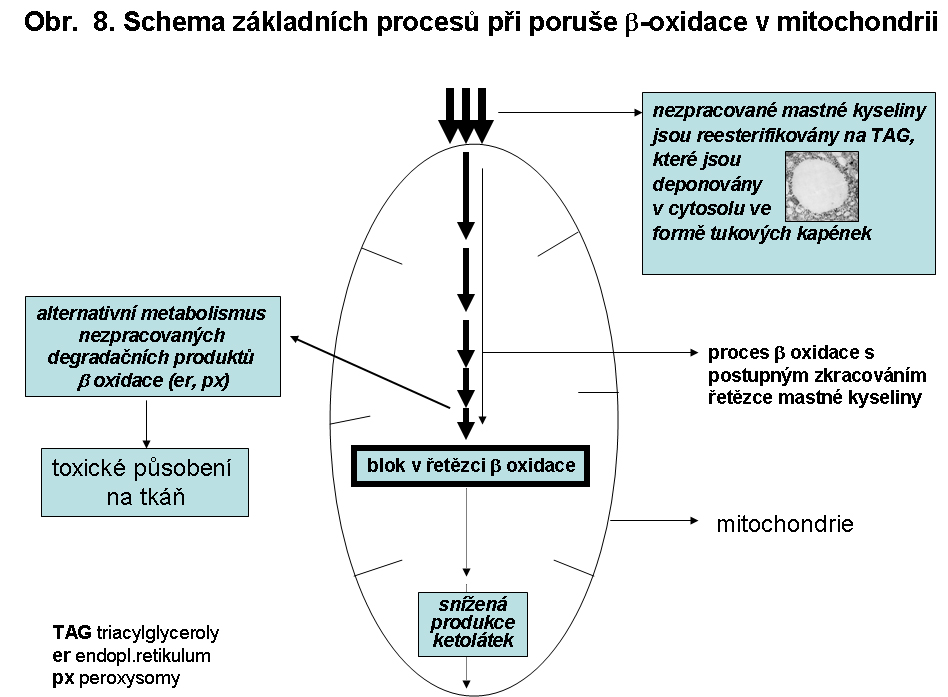
Porucha je míněna v nejširším slova smyslu, tj. zahrnuje vše před vlastním procesem oxidační spirály (transport mastné kyseliny do mitochondrie) a po ní (ketogenese)
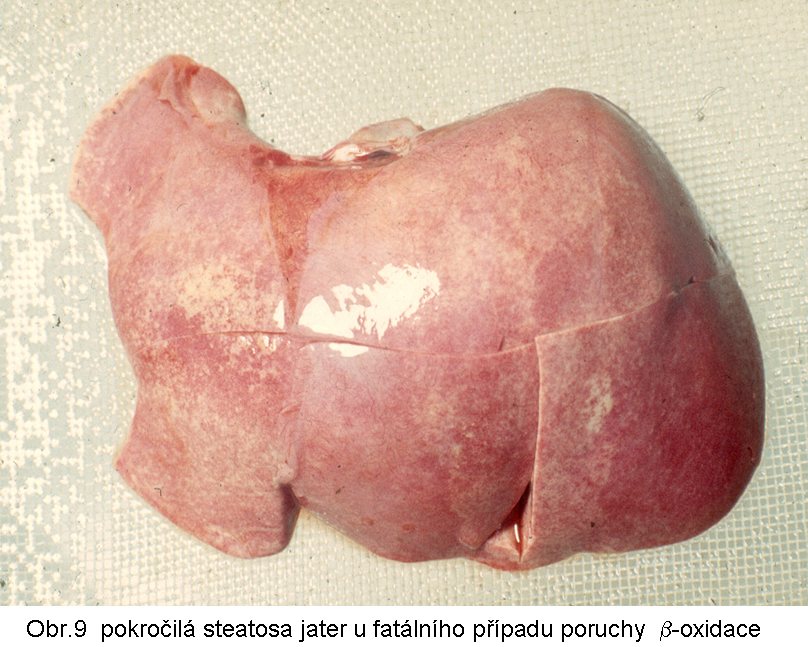
hepatopatologie: steatosa (obr.9), (obr.10), (obr.11), (obr.12), (obr.13), selhání jater, cirrhosa jater
kardiopatologie: steatosa, hypertrofická kardiomyopatie, endokardiální fibroelastosa, poruchy vodivého systému
neuropatologie: edém mozku, residuální změny po atakách mozkového edému, periferní neuropatie (senso-motorická), pigmentová retinitis (retinální pigmentový epitel je velmi citlivý na deficienci BOX)
myopatologie: klinicky obraz myopatie , často s atakami rhabdomylosy, histologicky steatosa (obr.15) (nemusí být vyznačena, nebo jen nevýrazně) takže mohou být přítomny pouze nespecifické změny.
Popisovány jsou i nefrologické poruchy (tubulopatie) až selhání ledvin; běžná je steatosa kanálkového epitelu (obr.14)
Relativní příznačnost patologického nálezu může být snížena minimálním stupněm steatosy
Klinický průběh poruch beta oxidace je často v atakách hypoketotických hypoglykémii, vyvolaných hladověním nebo infekcí, při kterých nastane mobilizace MK z tukové tkáně a jejich transport do jater a orgánů a tím k deficitním mitochondriím. Řada případů tzv. Reyova syndromu (hepatocerebrální syndrom charakterisovaný selháním jater a mozkovým edémem) nebo syndromu náhlých úmrtí je vysvětlitelná dědičnými poruchami beta oxidace. Toxické projevy těchto poruch, centrované na játra (jaterní selhání) a mozek (mozkový edém) jsou pravděpodobně způsobeny toxickými alternativními produkty poruchy beta oxidace. Značnou roli hraje i hypoglykémie, která je integrálním nálezem v atakách. V řadě případů je průběh kontinuální s případnými excarbacemi (viz shora). Orgánová manifestace je velmi různá. Kolísá od adultních myopatických forem s rekurentní rhabdomyolysou, nebo hypertrofickou kardiomyopatií po hydrops fetus
léčba : zvládnutí hypoglykémie
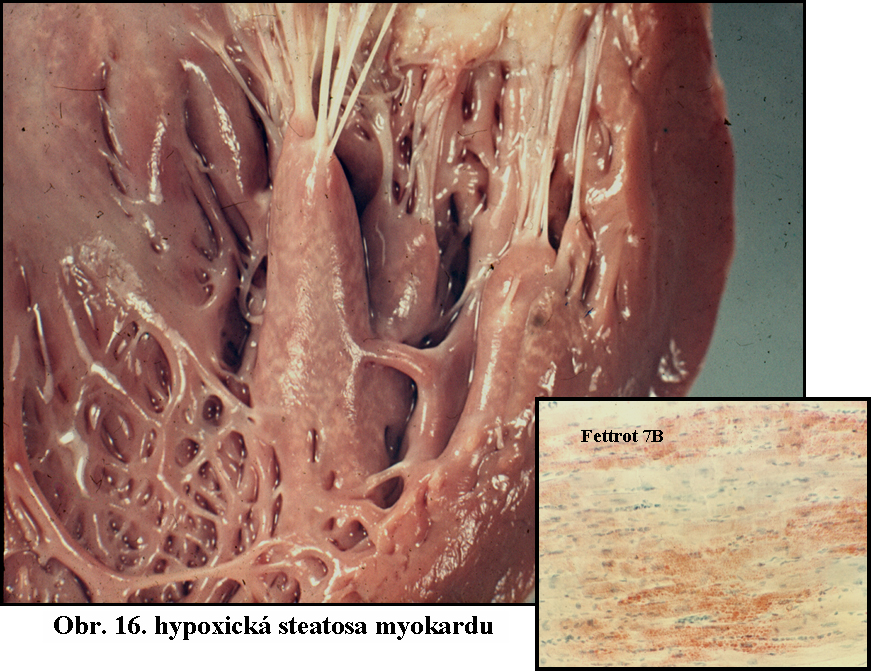
Získané stavy - zdaleka nejčastější příčinou je ischémie (nedokrevnost), hypoxie a anoxie. Ischémie myokardu je vždy provázena steatosou kardiocytů. Krajní stav ischémie - infarkt má vždy nažloutlou barvu díky výrazné steatose, která se v těžce ischemisovaných vláknech rozvíjí. Klasickým celkovým projevem hypoxie je obraz t.zv. tygrovaného srdce - nejčastěji následek výrazné anemie, vysvětlovaný jako akcentace hypoxie (a tedy steatosy) v okrscích srdečního svalu v blízkosti venosního konce kapiláry, kde došlo k výrazně snížené tensi kyslíku (obr.16). Se steatosou podmíněnou ischémii se lze setkat i dalších orgánech (okraje infarktů, tzv. "muškátová játra" u těžkých venostázách jaterních při selhání srdečním)
III. Degradace lipidů v lysosomech a její poruchyZa normálních okolností jsou lipidy v lysosomech hydrolyzovány na sfingosin, glycerol, mastné kyseliny, hexosy, sulfát a cholesterol sadou lysosomálních kyselých hydroláz (lipáz, fosfolipáz, glykosidáz, sulfatáz). Produkty difundují přes lysosomální membránu a jsou buňkou reutilisovány. Dokonalá funkce lysosomálního aparátu je prevencí lysosomální steatosy (obecně akumulace lipidů v lysosomech). Jedinou výjimkou je cholesterol, který je postlysosomálně reesterifikován a jeho kapky mohou být i po různě dlouhou dobu v cytosolu deponovány (viz dále).
Poznámka
V kapitole pojednávající o lysosomálním systému bude zmíněna o nedávno definované funkce p?enosu lipid? v lysosomálních membrán do dalších kompartment? bu?ky (lipid trafficking), jehož porucha vede k akumulaci nejr?zn?jších lipid? i p?es p?ítomnost p?íslušných hydrolás.
1. Získané stavy jsou spojeny s intensivní endocytosou nebo fagocytosou
(dřívější termín resorpční steatos; pojem resorpce sdružuje jak fagocytosu, tak endocytosu)
Za normálních okolností je endocytovaný, či fagocytovaný materiál kompletně hydrolyzován baterií lysosomálních hydroláz na základní molekuly, které jsou recyklovány po přestupu lysosomální membrány. Dočasně expandovaný systém lysosomálních membrán je redukován a převeden do původního, resp. klidového stavu.
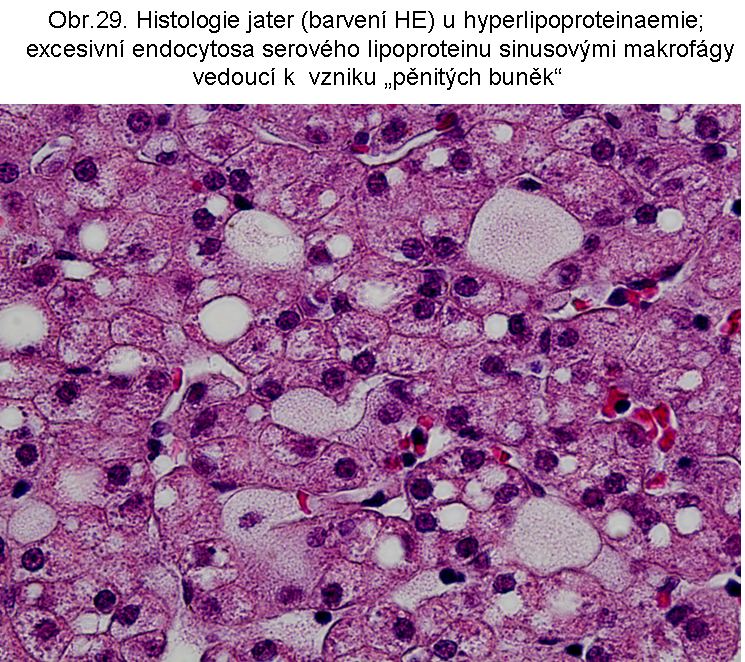
a. Za některých patologických okolností může dojít k parciální (relativní, dočasné) lysosomální nedostatečnosti, způsobené obzvláště intensivní endocytosou lipidů a to buď ve formě lipoproteinů nebo struktur bohatých na lipidy (destičky krevní, erytrocyty, myelin, ap.). Tato steatosa se tedy týká prakticky výlučně histiocytů, které se mění na pěnovité buňky (foam cells) s lysosomy vyplněnými lipidem, případně jiným materiálem (výjimky viz níže). Lipidy jsou tedy za tohoto stavu retinovány pro zpomalenou hydrolysu intralysosomálně (kapky jsou ohraničeny jednoduchou membránou). Jejich intralysosomální spektrum je určeno typem endocytovaného materiálu a časovým faktorem, neboť existují rozdíly v rychlosti hydrolysy jednotlivých lipidních typů.
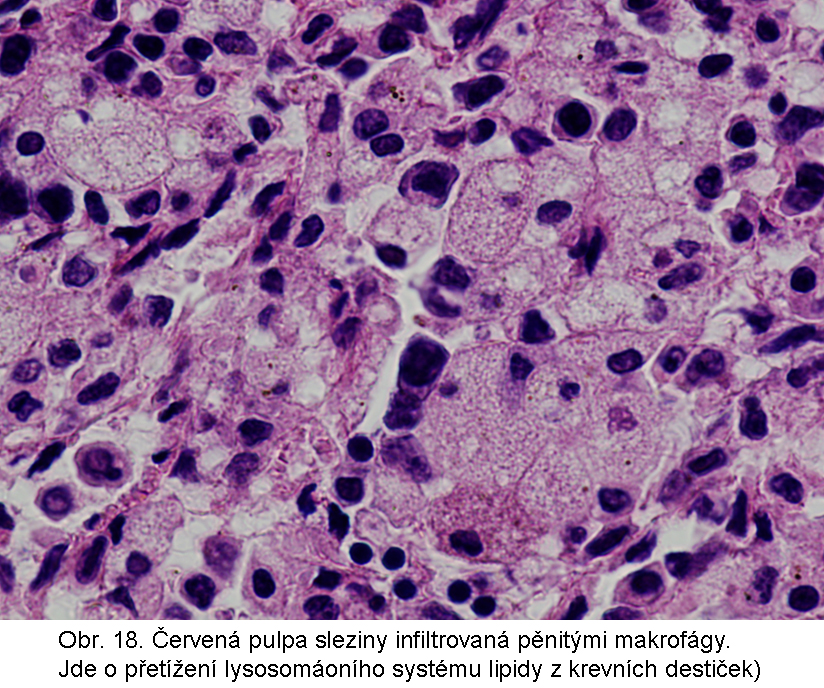
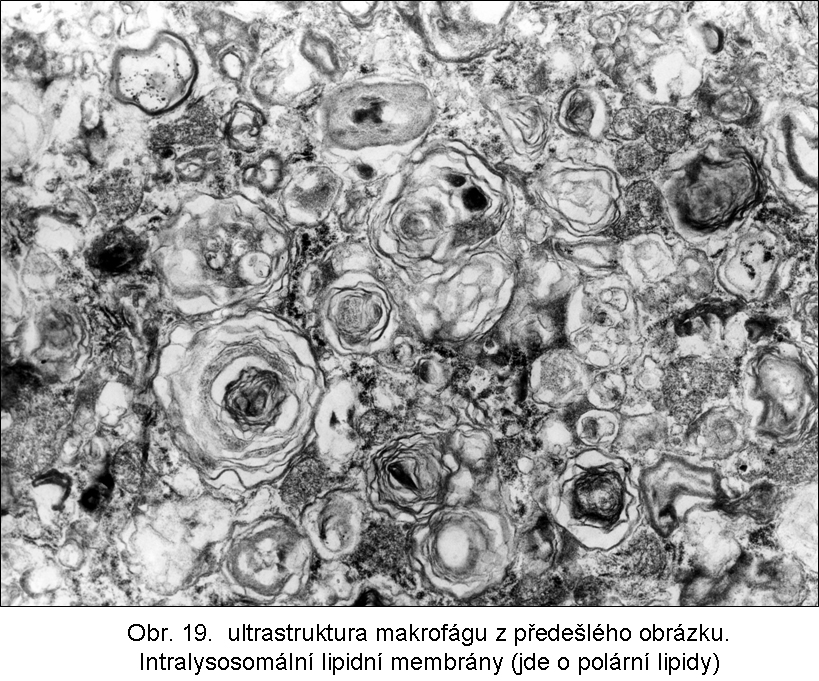
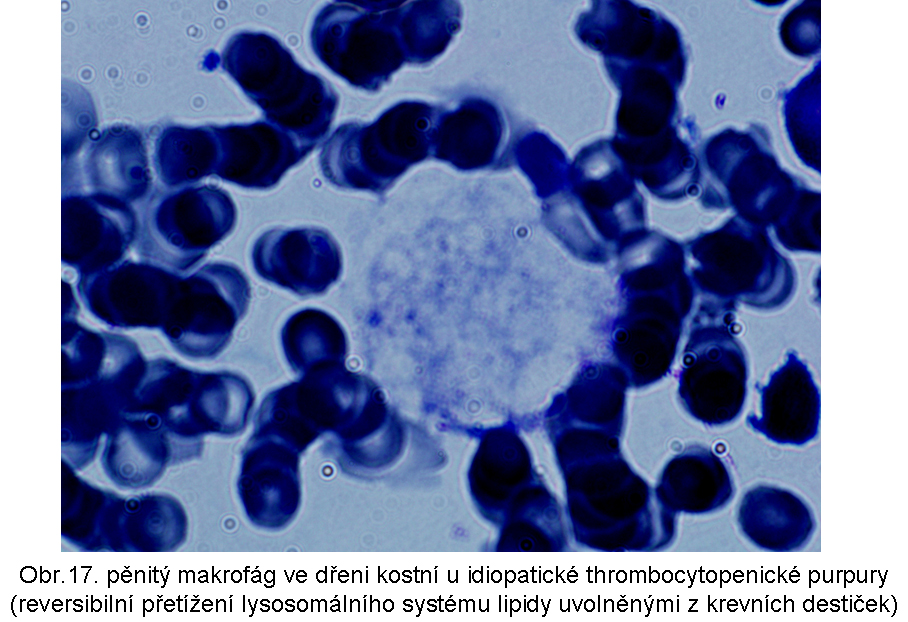
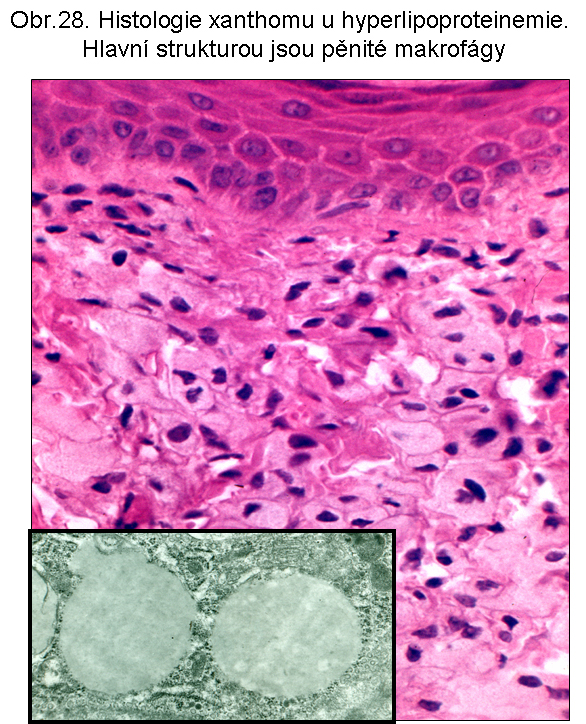
stavy: jde o stavy s deposicí pěnitých histiocytů (foam cell syndrom), který může rozsahem v některých případech připomínat primární lysosomální střádací proces. Může být přítomno různé množství lipopigmentu. Celý proces je lokalizován pouze v makrofázích - tedy bez postižení ostatních buněčných typů. Jeho maximum je ve slezině (obr.18), (obr.19), ale pěnité buňky mohou být i v játrech a zejména ve dřeni kostní (obr.17). Stavy při kterých k této situaci dochází: poruchy charakterizované nadměrnou fagocytosou trombocytů (idiopatická trombocytopenie) nebo lipoproteinů (dyslipoproteinemie, hyperlipoproteinemie). Znalost těchto stavů je důležitá pro efektivní diferenciální diagnosu primárních lysosomálních střádacích poruch.
b. excesivní převážně "postlysosomální" akumulace lipidu. Velmi časté jsou situace, kdy dominantním endocytovaným/fagocytovaným lipidem je cholesterol (nebo jeho estery). Po průniku do cytosolu je reesterifikován příslušnou acyltranferasou (cholesterolacyltransferasa v membránách endoplasmatického retikula) na ester a deponován ve formě kapénky esterů cholesterolu bez limitující membrány (postlysosomální depo). Jeho další osud závisí na další utilisaci buňkou, což je v případě steroidy neprodukujících buněk obtížné. V těchto buňkách je zřejmě kruciálním momentem extrakce cholesterolu pomocí plasmatických HDL (vysoce densních lipoproteinů).
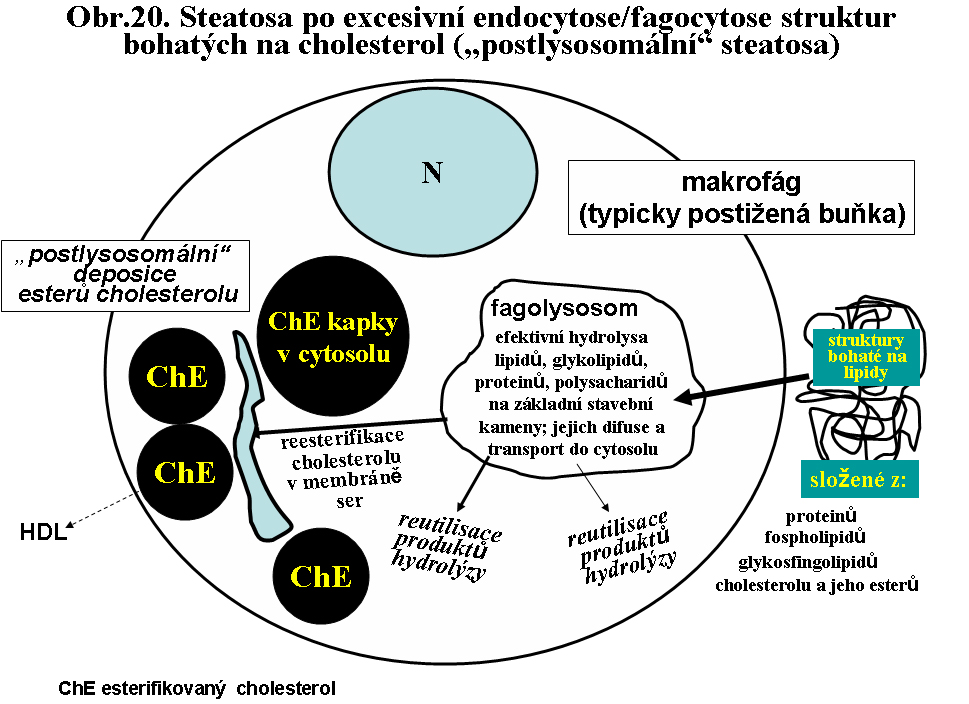
Za těchto stavů dochází k nestabilitě myelinu, který je fagocytován mikrogliálními fagocyty. V jejich fagolysosomech jsou veškeré komponenty myelinu efektivně degradovány na jednotlivé stavební kameny, které difundují (nebo jsou aktivně transportovány) přes lysosomální membránu do cytosolu, kde jsou použity pro další procesy a tedy nedochází k jejich akumulaci. Jedinou výjimkou je cholesterol, který, pokud je akumulován v lysosomech je transportován (doposud ne zcela definovaným mechanismem; významnou roli zřejmě hraji NPC proteiny) do membrán endoplasmatického retikula, kde je esterifikován (viz shora ACAT - acyl-Koenzym A:cholesterol acyltransferasa) a deponován ve formě tukové kapky v cytosolu (viz shora biologie tukové kapky). Výsledkem je tedy "postlysosomální" akumulace esterů cholesterolu ve formě kapének, které se barví sudanovými barvivy jako granula - zrnéčka. Odtud historický název zrnéčkové buňky (obr.21)
Pokud nejde o deficit lysosomálního enzymu štěpícího některý ze sfingolipidů myelinu (arylsulfatasy A - deficitní u metachromatické leukodystrofie nebo galaktocerenrosidasy, deficitní u Krabbeho nemoci) dochází k hromadění toho lipid jako projev lysosomálního střádání na podkladě enzymopatie. Nutno podotknout, že k akumulaci esterů cholesterolu v detekovatelném množství nedochází
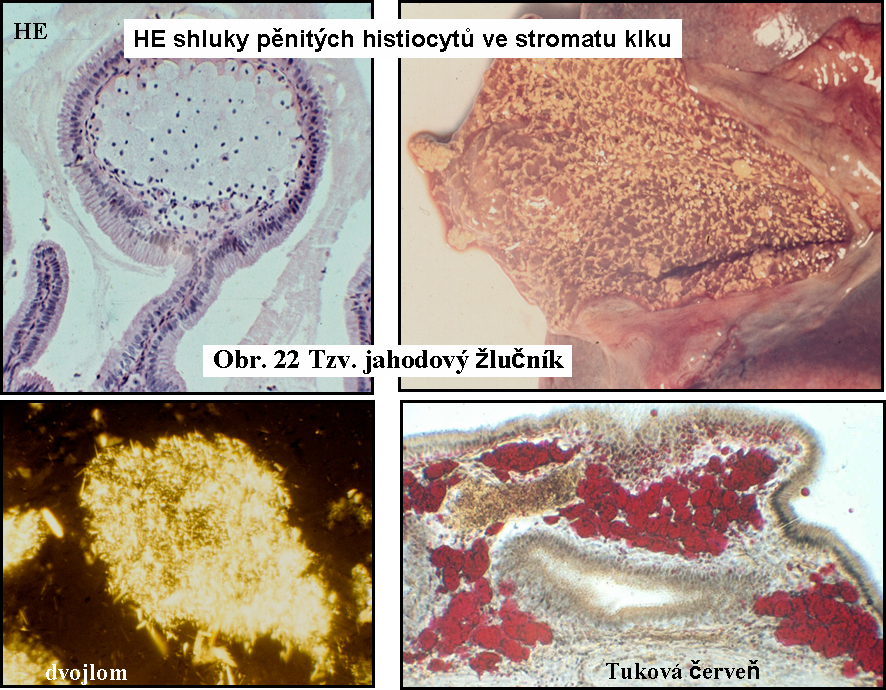
Lipidní fagocyty v dalších tkáníchLipidní fagocyty nabité cytosolárními kapkami esterů cholesterolu jsou známé v řadě dalších stavů, kdy dochází k zvýšené fagocytose lipidů s příměsí esterů cholesterolu, nebo volného cholesterolu, např. ve sliznici žlučníku (cholesterolosa žlučníku) (obr.22), v chronických abscesech (pseudoxanthom), v aterosklerotických lesích. Lipidní fagocyty ve žlučníku jsou výrazem endocytosy žluče bohaté cholesterolem.
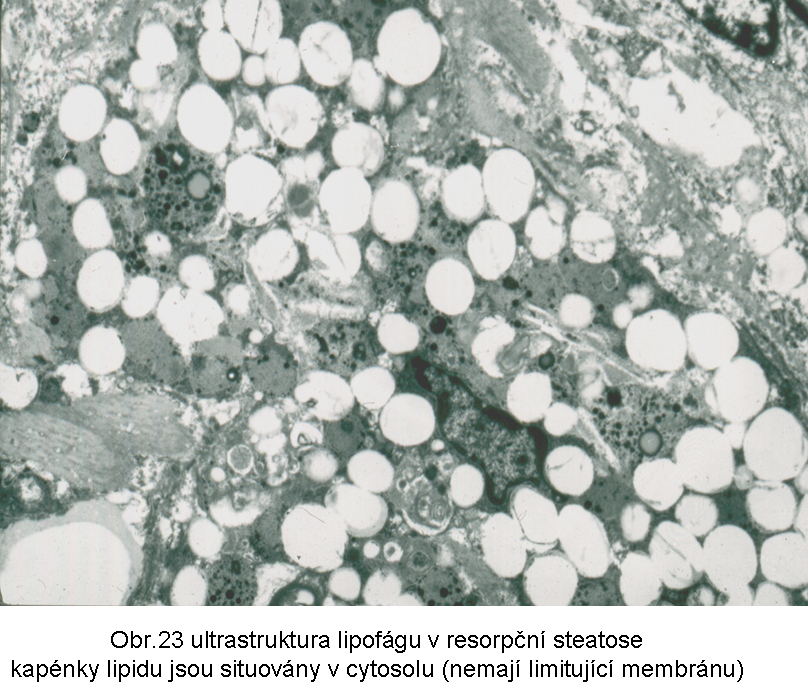
Přesto, že jde o akumulaci esterů cholesterolu v cytosolu (obr.23), mohou být přítomny známky aktivovaného lysosomálního systému, který hraje centrální roli v tomto mechanismu steatosy. Nemůže být překvapením, že mohou být v lipidních fagocytech detekovatelné lysosomy obsahující lipid nebo jiné látky, pocházející z fagocytovaného materiálu. U všech těchto stavů může být i sekundárně zmnožen paralelní intralysosomální produkt - lipopigment typu ceroidu, zvláště dojde-li k prodloužení fáze intralysosomální digesce. Důvodem je přetížení lysosomálního systému.
poznámky. I jiné buňky s výraznou endocytickou schopností mohou steatosu tohoto typu vykazovat. Jde zejména o hepatocyty (intensivní endocytosa lipoproteinů) nebo buňky proximálních kanálků ledvin (endocytosa lipoproteinů při patologické propustnosti glomerulárních basálních membrán).
Analogická situace jako ve shora zmíněných situacích (charakterisovaných akumulací esterů cholesterolu) může nastat s TAG, i když tyto situace jsou výjimečné (fagocytosa málo densních lipoproteinů nebo tukové tkáně).Může dojít i ke kombinované akumulaci ChE i TAG, podle situace (např. v atherosklerotických plátech)
Dle ustálených zvyklostí jsou dědičné enzymopatie nazývány lipidosami, nebo onemocnění střádací. Termín střádací je dán biologickou výjimečností této skupiny, protože akumulovaný substrát se hromadí v uzavřeném kompartmentu deficitního enzymu, který se následkem toho expanduje a podmiňuje proces zvětšování buňky a následně i orgánu. Detailně jsou tyto poruchy probrány v části pojednávající o fysiologii a patologii lysosomálních střádacích poruch. Již zde je vhodné připomenou, že tyto genetické poruchy jsou dány velkou skupinou enzymopatií a menší, ale velmi významnou skupinou deficitu nekatalytických proteinů, které zajišťují transport z lysosomu do cytosolu, transport membránových lipidů buňkou nebo representují funkce, které nebyly doposud definovány
Další mechanismy vedoucí k deposici lipidů v buňceV případě zvýšeného přísunu mastných kyselin z plasmy, klasicky při mobilizaci z tukové tkáně, např. při hladovění, nebo u nedostatečné utilisovatelnosti cukrů; jde o kombinaci nedostatečně rychlé mitochondriální utilizace s nedostatečnou asemblaci na lipoproteiny a jejich sekrecí. V obou případech se z nadbytečných mastných kyselin vytvoří depo TAG, jehož drenáž je zřejmě možná zvýšením sekretorického procesu (pokud by byla vystupňována transkripce příslušných apoproteinů. Játra v tomto smyslu slouží jako reservní depo (viz úvod kapitoly).
Deficitní extrakce cholesterolu z buněkTangierská nemoc. Jde o deficit transportu cholesterolu z buněk, podmíněný mutací ABCA1 transportéru cholesterolu. Tento na sebe váže HDL apolipoprotein A-I ve stavu, kdy obsahuje malé množství lipidů, předá cholesterol, který obohacuje partikuly lipoproteinu a je transportován do jater (RTC reverse cholesterol transport). Pokud je tento transportér mutován je transport cholesterolu z buněk inhibován, cholesterol v esterifikované formě je retinován v cytosolu buněk a přeměňuje je na buňky pěnité, srovnatelné s "postlysosomální akumulací" u excesu endocytosy (viz shora). Cholesterol je retinován v makrofázích (splenomegalie, obrovské žluté tonsily, sinusové buňky jater, atd), ve Schwannových buňkách (neuropatie),ale ne v dalších buněčných typech, ve kterých se tedy nedostatečná extrakce cholesterolu zřejmě musí manifestovat jiným způsobem než jeho retencí v esterifikované formě v cytosolu. Jelikož lysosomální systém není při této poruše ve hře, není tendence k tvorbě ceroidu, i když trvání poruchy je velmi dlouhé. Nedostatečná "lipidisace" HDL apoproteinů vede k jejich akcelerované degradaci, což vysvětluje jejich nízkou hladinu v plasmě
Poruchy hydrolýzy TAG v cytosolárních kapkách (viz shora biologie tukové kapky)Chanarin-Dorfmanův syndrom ( je charakterizován prakticky generalizovanou deposicí TAG v cytosolu nejrůznějších typů buněk: keratinocytů, Langerhansových buněk, buněk hladkého svalu, dermálních fibroblastů, Schwannových buněk, ve vláknech kosterního svalu, v enterocytech, z epitelu žaludeční sliznice, v granulocytech a monocytech. Velmi postižené jsou hepatocyty (hepatomegalie, steatoheptitis), epidermis (ichtiosa, erythrodermie). Pro dg. je důležitý fenomén steatosy periferních granulocytů (Jordanova anomálie - přítomna údajně i u nosičů poruchy) a keratinocytů v biopsii kůže. Hladiny krevních lipidů nejsou změněné. Jde o geneticky podmíněnou poruchu autosomálně recesivního typu, jejíž podstatou je mutace CGI-58 proteinu (ABHD5, který se váže na lipázu štěpící TAG a umožňuje její aktivitu (viz shora). Porušena je degradace TAG cytosolární tukové kapky (viz shora). Mechanismus ichtiosy není uspokojivě vysvětlený. Střádání neutrálních lipidů (neutral lipid storage disease) je porucha velmi blízká Chanarin-Dorfmanovu syndromu tím, že jde stejně tak o akumulaci TAG v cytosolu buněk, ale na podkladě deficitu samotné lipázy štěpící cytosolární TAG (PNPLA2, zvaná též ATAGL, viz shora). Ve fenotypu chybí ichtiosa
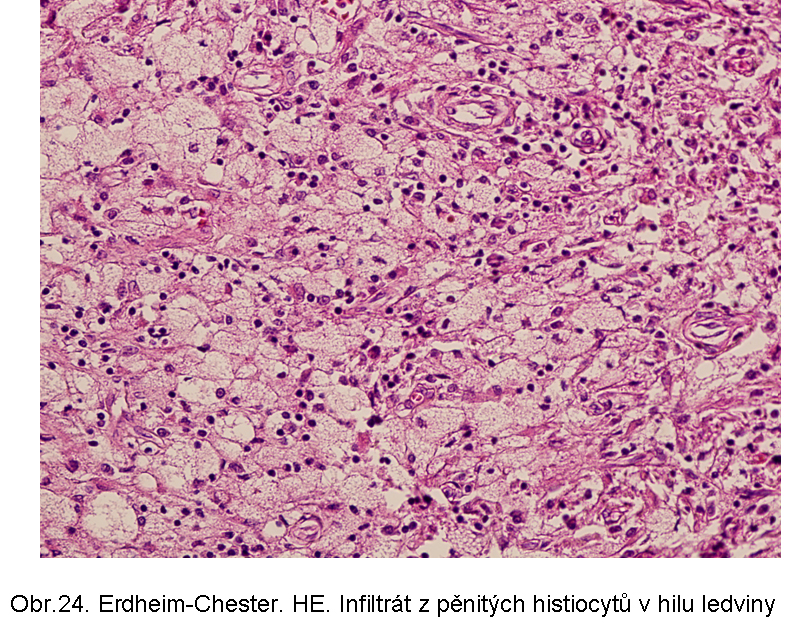
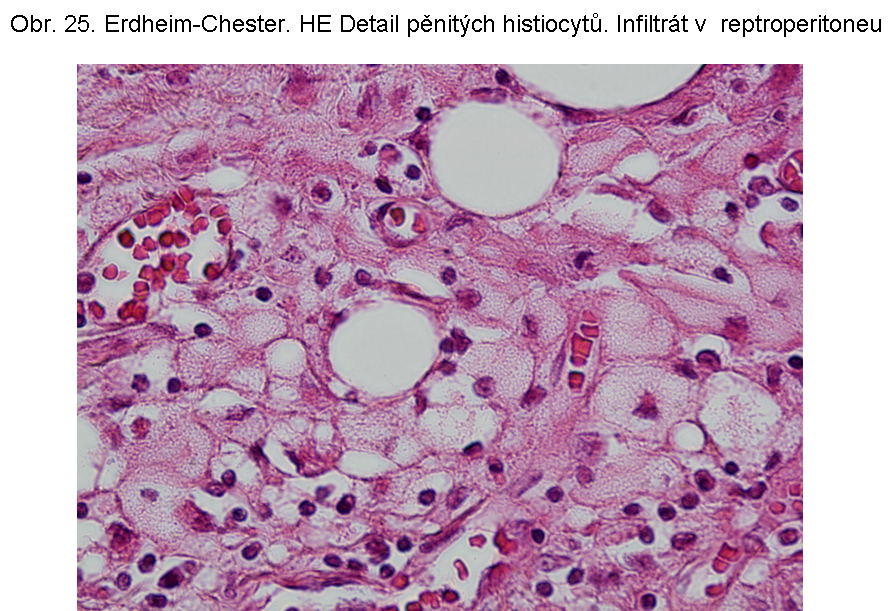
Patogeneticky nejasné akumulace esterů cholesterolu z buněkErdheimova Chesterova nemoc je charakterizována popisně jako histiocytární infiltráty z pěnovitých histiocytů s cytosolárními kapénkami převážně z esterů cholesterolu (odtud název xanthogranulomatosní lése). Průvodní složkou je fibroplasie. Jak řečeno, dominuje extralysosomální akumulace lipidu. Proto je deposice ceroidu spíše okrajové, nebo výjimečná. Příčina akumulace lipidu v makrofázích není jasná. Ložiska se vyskytují zejména v kostech, plicích, retroperitoneu (obr.24, 25), ale i dalších lokalizacích, včetně CNS.
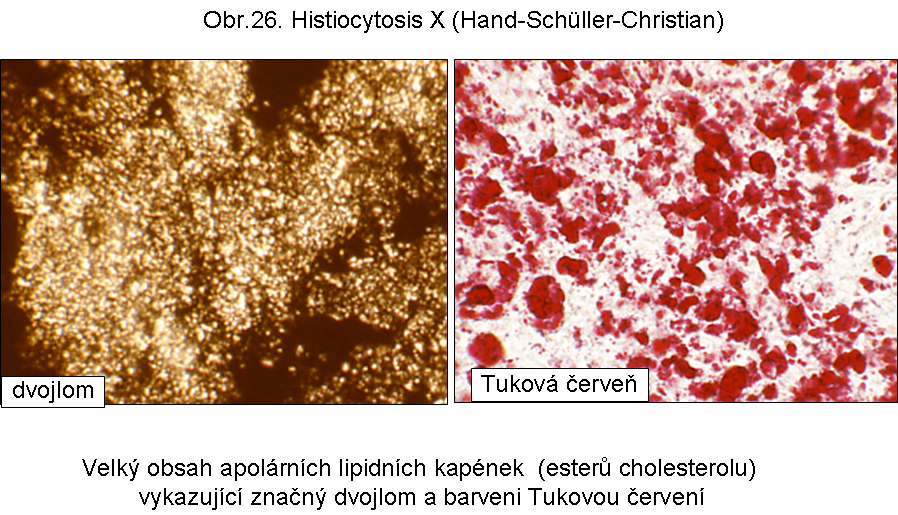
Analogická patogeneticky nejasná cytosolární akumulace esterů cholesterolu je v Langerhansových buňkách v rámci Hand-Schueller-Christianovy histiocytosis X. (obr.26, 27)
Steatosy dané komplexními mechanismyBereme-li krevní systém jako samostatný kompartment, pak nutno považovat tzv. hyperlipemie za analogii steatosy (extracelulární). Problém mechanismu vzniku hyperlipoproiteinemií, ev. dyslipoproteinemií je natolik specielní, že se vymyká zaměření této kapitoly, která je zaměřena na jejich následky.
Část lipoproteinu může být odstraněna nespecifickou endocytosou histiocytárními elementy. Experimentálně bylo dokázáno, že po modifikaci lipoproteinů (např. oxidací, modifikací lysinových zbytků) se vytvářejí patologické ligandy, pro které mají histiocyty receptory, což má za následek zvýšenou resorpci lipoproteinu cestou receptorově zprostředkované endocytosy. Oxidované lipoproteiny však mohou být toxické pro celou řadu buněk, zejména pro endotelie.
Zvýšená hladina lipoprotein? má za následek n?kolik velmi závažných komplikací:Jde o ty stavy, za kterých dochází k nadměrné akumulaci abnormálních lipidů, např. prekursorů cholesterolu nebo abnormálních mastných kyselin v lipidech buněčných membrán. Tím jsou silně narušeny fyzikální vlastnosti membrán a dochází k regresi buněk, aniž by docházelo k vytvoření patologické samostatné lipidní fáze ve formě kapének, jako u shora zmíněných steatos.
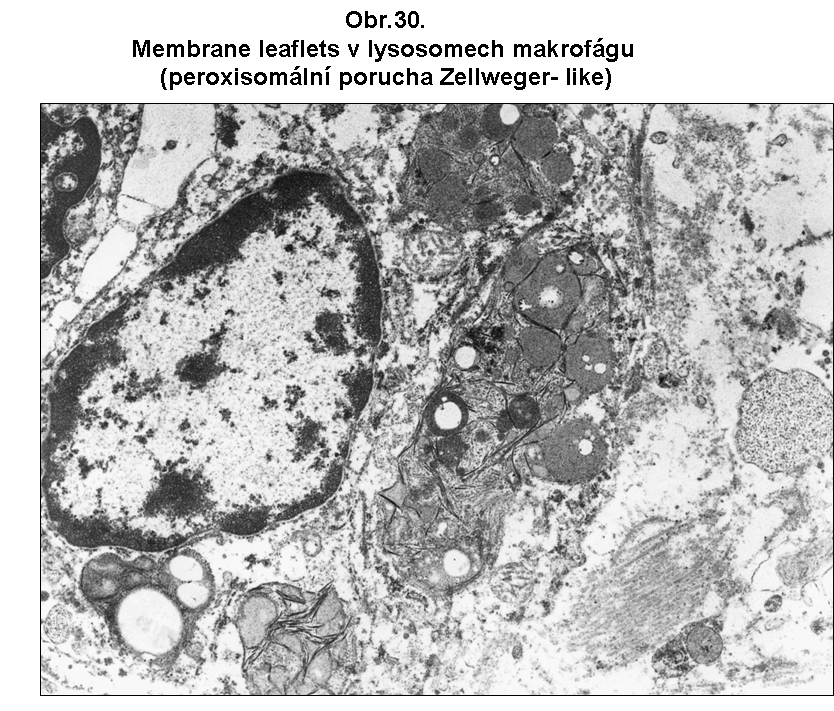
Klasicky je tomu u defektů peroxisomální oxidace mastných kyselin s velmi dlouhým řetězcem (C26:0, C:24 - jejich koncentrace a poměry mezi nimi a C:22 jsou dány v příslušných příručkách klinické biochemie). Nedostatečná peroxisomální oxidace vede k nadměrné akumulaci těchto mastných kyselin s velmi dlouhým řetězcem a k jejich včleňování do membránových lipidů nejrůznějšího druhu. Nelze vyloučit ani nadměrnou acylaci proteinů akumulovanými mastnými kyselinami za těchto stavů. Podle současných představ toto vede k abnormálním fyzikálně chemickým vlastnostem membrán a ke snížené vitalitě buněk. Jde tedy o postižení mnohem významnější než u klasické steatosy (viz výše). Za těchto stavů dochází k akumulaci krystalků esterů těchto dlouhých mastných kyselin s cholesterolem v cytoplasmě. Nejasná je také povaha zvláštních štíhlých lístkovitých membránových formací, které jsou některými autory považovány za místo akumulace velmi dlouhých mastných kyselin. Jsou však zcela resistentní i na nejnásilnější způsoby extrakce. Nejvíce postižené jsou kortikální epitelie nadledviny, Leydigovy buňky a myelin, který se stává nestabilním a je odbouráván, což vede k leukodystrofii Vzhled postižených buněk je necharakteristický. V cytologickém obraze mohou dominovat lístkovité membránové inkluse lokalisované velmi často v lysosomech (obr. 30). Povaha těchto formací je neznámá; vzdorují nejagresivnějším lipidním extrakcím.
Analogický stav existuje u cerebrotendinosní xanthomatosy, způsobené poruchou syntesy žlučových kyselin v játrech. Akumulovaný intermediární produkt - cholestanol - se inkorporuje do buněčných membrán, zejména do myelinu, což vede k jeho nestabilitě a rozpadu.
Smith-Lemli-Opitz (SLO). Jde o defekt v terminální fázi syntesy cholesterolu na podkladě deficitu 3β hydroxysteroid Δ7 reduktázy). Předpokládá se, že pravděpodobně vede, zatím ne zcela jasným způsobem, ke změnám lipidních membránových mikrodomén. V nich hraje přítomnost cholesterolu velmi významnou roli, která může být narušena interferencí 7 dehydrocholesterolu
Dosáhne-li steatosa značného stupně, je patrná i makroskopicky. Postižené orgány jsou žluté (v případě nepolárních steatos - příčinou je pravděpodobně jak stupeň nenasycenosti lipidu, tak příměs karotenoidů). Barva může být modifikována stupněm překrvení (směr do oranžova). Barva tzv. hnědé tukové tkáně je dána jednak bohatostí cév, jednak vysokou koncentrací cytochromů v početných mitochondriích. V případě excesivní akumulace polárních lipidů je barva bílá (myelin) nebo šedobílá.
Fysikální stav deponovaného lipidu - dvojlomObě základní skupiny akumulovaného lipidu u steatos se liší fysikálním stavem. TAG jsou přítomny tekuté formě, tj. nemají dvojlom(nebo dvojlom zcela rudimentární). Naproti tomu kapky esterů cholesterolu mají charakter tekutých krystalků a vykazují fenomén maltézského kříže (podobně jako řada polárních lipidů - tyto jsou však resistentní na extrakci bezvodým acetonem, která odstraní deposita esterů cholesterolu). Tento stav se může měnit na pevnou krystalickou fázi (krystalky esterů cholesterolu)
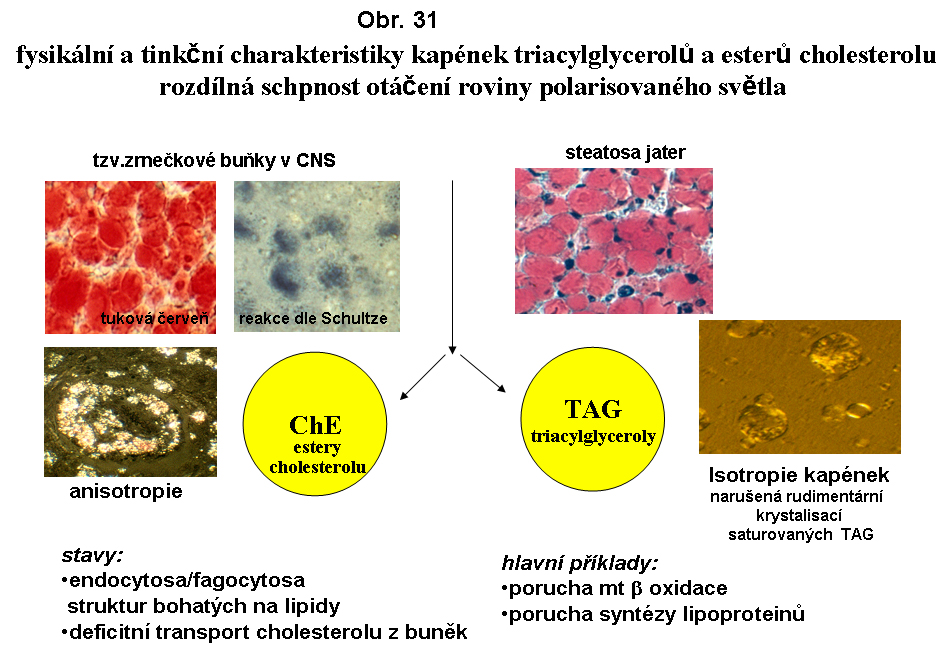
termín steatosa (ztukovění) - je reservován pro poruchy získané. Mikroskopickým projevem steatosy je intracelulární tuková kapénka lokalizovaná v cytosolu. V těchto případech vždy o nepolární, silně hydrofobní lipidy. Stupeň agregace závisí pravděpodobně na kvalitě hraniční vrstvy (viz shora biologie tukové kapky). Faktorem působícím proti agregaci mohou být látky adsorbované na hraniční zoně lipidní kapky (polární lipidy, proteiny). Extremním stavem mnohočetné (multilokulární) vakuolisace je tzv. pěnitá (angl. foamy) struktura cytoplasmy. Extremním stavem kumulace silně agregujícího lipidu je solitární megavakuola (monovakuolární, unilokulární steatosa, tuková cysta) distendující cytoplasmu. Tento stav může vést až k ruptuře buňky. Klasicky je tomu v případě steatosy hepatocytů. Regulovaná unilokulární distense je fyziologická u buněk bílé tukové tkáně.
termín lipidosa je reservován pro geneticky podmíněné poruchy lysosomální degradace lipidů nejrůznějšího druhu. Jde tedy o přeplňování lysosomálního systému nedegradovaným lipidem, který vede k expansi lysosomálního systému a k tím k výrazným cytologickým změnám postižené buňky.
Vitalita a další osud buňky postižené steatosouSamotná akumulace tuku, ať už z jakýchkoliv příčin, neovlivňuje hostitelskou buňku prokazatelně výrazněji negativně. Prognostickým faktorem je samotný vyvolávající faktor (jeho další účinky) nebo deficit v utilisaci akumulovaného lipidu (klasicky u alternativního zpracování retinovaných mastných kyselin u poruch BOX - viz shora). Lze předpokládat, že jedině excesivní akumulace lipidu vede k biologické deterioraci a nekrose postižené buňky, a to ať už se jedná o nadměrné hromadění v cytosolu nebo v lysosomech.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}