Seznam zkratek: AGL - amylo 1,6-glukosidasa, 4-a glukantransferasa; AMPK - AMP aktivovaná protein kinasa; APGBD - adult polyglukosan body disease; CA -corpora amylacea; G6P - glukosa-6-fosfát; GBE - glycogen brancher enzyme; GDE - glycogen debrancher enzyme; GS - glykogen synthasa; GSD - glycogen storage disorders; GSK - glykogen synthasa kinasa; KMP - kardiomyopatie; PH - fosforylasa (phosphorylase); PRK - fosforylasa kinasa (phosphorylase kinase); PTG - protein targeting to glycogen; SGLT - sodium glucose transporter; SRC - steroid receptor coactivator; UDPG - uridin difosfát glukosa
Glykogen je lineární a bohatě větvený polymer (dendrimer) složený z molekul α-D glukosy. Patří mezi glukany. Je to základní zásobní polysacharid živočichů. Větvení glykogenu prostřednictvím α1-6 glykosidové vazby se vyskytuje po podle některých po 3-7, podle jiných po 12-14 lineárních glukosových jednotkách, spojených vazbou α1-4. Jedna molekula glykogenu má mezi 106- 107 daltonů a odhaduje se, že obsahuje okolo 60 000 molekul glukosy. Obsahuje vždy jisté množství estericky vázané kyseliny fosforečné, která spolupodmiňuje jeho velkou bobtnavost. S jodem dává glykogen hnědé zbarvení. Archaicky bývá někdy nazýván živočišný škrob.
V říši rostlinné je analogem glykogenu škrob, patřící mezi větvené polysacharidy rostlinné buňky. Škrob je nejdůležitějším zdrojem sacharidů ve výživě člověka. Na rozdíl od glykogenu je škrob za studena nerozpustný. Škrob není chemické individuum. Skládá ze dvou složek: minoritní složkou (15-20%) je lineární amylosa skládající se zhruba z 1000 glukosových monomerů pospojovaných α1-4 vazbami. Polymer se stáčí do levotočivé šroubovice, dostane-li se do ní jod, vzniká modré zbarvení. Hovoří se o vytvoření inklusní komponenty. Majoritní složkou škrobu (80-85%) je větvený amylopektin. Větvení nastává tvorbou glykosidové vazby α1-6 v intervalu 20-24 glukosových jednotek lineárně spojených α1-4 glykosidovými vazbami. Stupeň větvení je tedy podstatně nižší než je tomu u glykogenu. Amylopektin s jodem dává červené zbarvení.
Větvení glykogenu je výhodné pro metabolismus a biologii buňky ze dvou důvodů. (i) zajišťuje solubilitu polymeru v cytoplasmě, (ii) umožňuje rychlou efektivní mobilisaci glukosy fosforylasou, která může zahájit degradační proces pouze z neredukujícího konce (terminální molekuly glukosy na konci větvení, mohou představovat 7 až 10% z celkového počtu)
V α-D glukanovém polymeru jsou údajně zabudována malá množství dalších cukrů (glykosaminů). Význam tohoto uspořádání není znám.
Glykogen lze natrávit amylasami. α amylasa štěpí glykogen uvnitř na větší fragmenty, které štěpí dále až na maltosu, případně glukosu; β amylasa je exoenzym odštěpující disacharidové jednotky z neredukujících konců, γ amylasa jediná štěpí 1-4 i 1-6 vazby a vyžaduje kyselé optimum - je identická s lysosomální α glukosidasou.
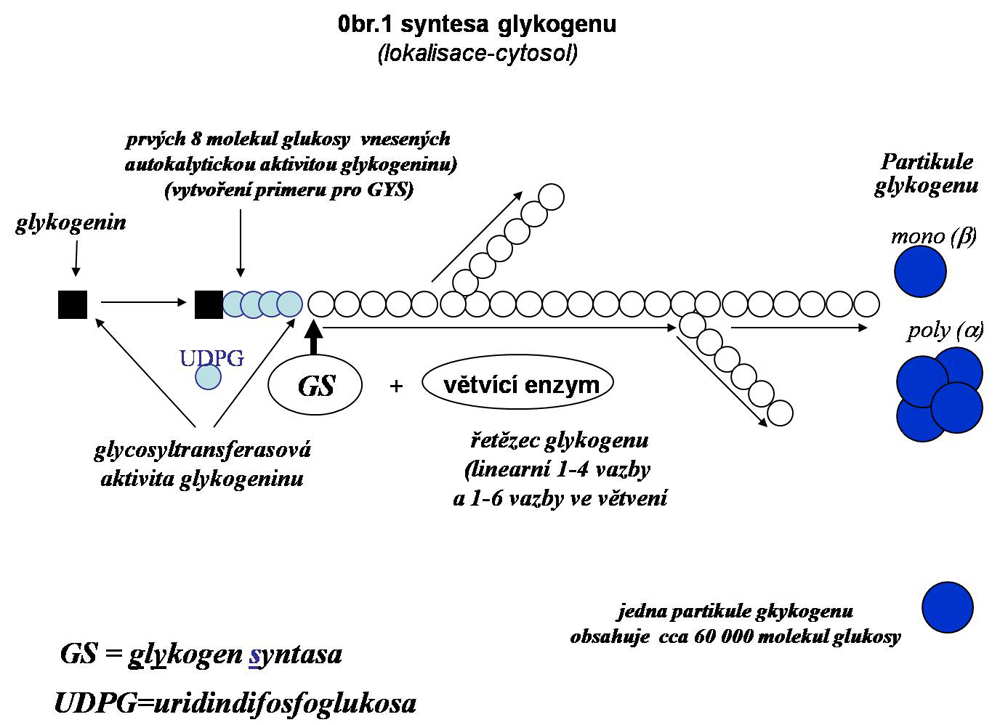
Synthesa glykogenu je zprostředkována glykogen synthasou (GS) a větvícím enzymem ("glycogen brancher" enzyme, GBE). Při všech stavech charakterizovaných zmnožením glykogenu (viz. níže) musí dojít k vytváření nových partikulí, neboť syntetická kapacita jedné partikule je regulačně omezena. Synthesa každé partikule začíná na molekule proteinu zvaného glykogenin. U primátů (na rozdíl od nižších vývojových druhů) jsou dva typy glykogeninu, kódované odlišnými geny. Glykogenin 1 (tzv. svalový, méně exprimovaný v jiných tkáních, např. v CNS) a glykogenin 2 (jaterní, ale exprimovaný i v srdci a v pankreatu). Glykogenin, působící nejspíše jako homodimer, má autokatalytickou kapacitu a katalysuje přenos molekuly glukosy na sebe z UDPG. Teprve po této iniciaci (vytvoření primeru) může nastoupit glykogen syntasa, která v souhře s větvícím enzymem vytváří rozvětvený polymer glykogenu (viz. obr.1) do určité (regulované) velikosti. β partikule mají konstantní průměr 20-30 nm a mají prakticky stejnou velikost jako ribosomy. Faktory regulující velikost granula glykogenu a faktory regulující jeho agregaci do α roset (viz. níže) nejsou známé.
Glykogen synthasa (GS), monomer, je regulována kinasovou aktivitou glykogen sythasy kinasy 3 (GSK 3), která fosforylací aktivitu enzymu inhibuje a která je v souhře se skupinou fosfatas (representovaných protein fosfatasou 1), které ji defosforylují (aktivují). Jsou známy dvě isoformy GS - jaterní (GS 2), exprimovaná pouze v játrech a svalová (GS 1) exprimovaná i v dalších tkáních). Obě kódované odlišnými geny. Významnou aktivací je allosterická vazba glukosa-6 fosfátu, která aktivuje protein fosfatasu 1 a tím aktivaci GS (synthesy glykogenu). Aktivaci GS zvyšuje úměrně i koncentrace glukosy (pravděpodobně přes zvýšenou tvorbu G6P)
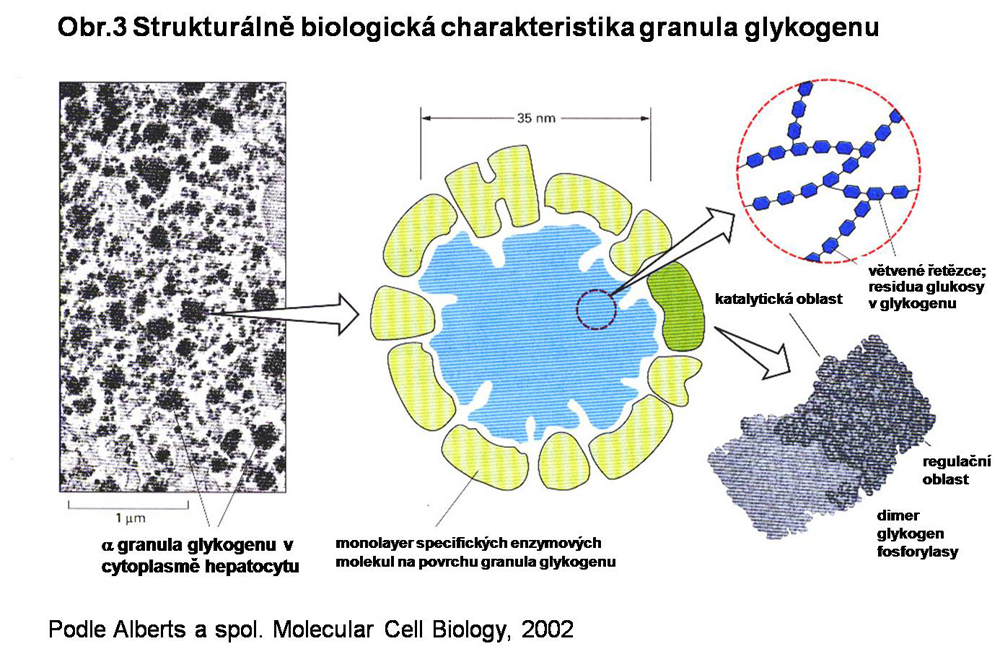
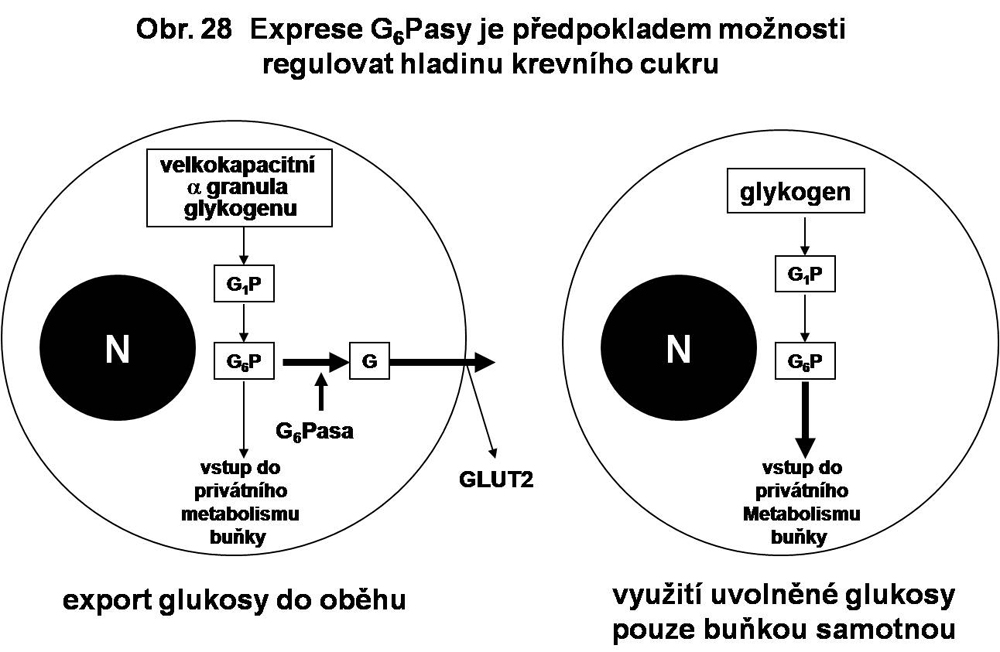
V některých buňkách existují mechanismy regulující stupeň agregace několika desítek β partikulí (20-30 nm) na větší rosetovité α agregáty o průměru 110-150 nm (viz. výše). Biologický význam dvojího typu uspořádání není jasný, je však známo, že v určitých tkáních je výskyt jednoho z typů granul typický, např. v hepatocytu je dominantní formou α částice, zatímco ve svalové tkáni je to β částice (viz. obr. 2). Zároveň platí, že zatímco hepatocyt (méně některé další buňky, viz. níže) dokáže přijímat glukosu, vytvářet glykogenová granula, ta štěpit, a glukosu uvolňovat z buněk k regulaci její hladiny v krvi, ve všech ostatních buňkách v těle, vzhledem k absenci glukoso-6-fosfatasy, může být glukosa utilisována pouze pro pokrytí vlastních energetických nároků.
Lze předpokládat, že α částice představují větší reservoár glukosy, což vysvětluje jejich standardní přítomnost v buňkách regulujících hladinu krevního cukru. Na povrchu partikulí jsou uloženy hlavní regulační enzymy obratu glykogenu: glykogen synthasa, glykogen synthasa kinasa (typ 3), komplex fosforylasy a její kinasy, fosfoprotein fosfatasa (typ1), včetně proteinů, které zajišťují adhesi zmíněných regulačních proteinů ke granulu glykogenu (viz. obr.3). Existují názory, že k iniciaci tvorby partikule glykogenu dochází v určitých predisponovaných oblastech cytosolu v úzké návaznosti na buněčné membrány
Je tendence nazývat glykogen o nízkém obsahu glukosy jako proglykogen, zralý glykogen jako makroglykogen. Někteří autoři definují i tzv. γ partikule glykogenu jako minipartikule o průměru okolo 3nm a to jak miniformy monopartikulátní ( β) tak rosetovité ( α). Existuje názor, že glykogen je čistě chemický pojem. Jelikož je v tkáním asociován s celou řadou proteinů, regulujících jeho obrat je doporučen termín glykosom (tento termín v některými autory používán pro agregáty glykogenu v lysosomech, ale i mimo lysosomy; jde o překlad termínu z anglosaské literatury; v úvahu připadá i termín glykogenosom) Pokud jsou glykogenová granula v cytosolu, je glykogen solubilní v kyselině (lyoglykogen, lyoglykosomy), pod je asociován s membránami, vzdoruje solubilisaci a tedy extrakci kyselinou (desmoglykogen, desmoglykosomy)
PoznámkyNeexistují doposud doklady pro přítomnosti solubilního (neagregovaného) glykogenu v buňkách. Málo známá je absence glykogenu v normálních neuronech, které mají jinak veškeré biochemické vybavení pro synthesu glykogenu, která je však za normálních okolností tlumena. Údajně má aktivní synthesa glykogenu za následek zánik neuronu apoptosou. Glykogen je tak považován za "Trojského koně" neuronu (viz. níže Laforova nemoc).
Glykogen persistuje v buňkách i po regulovaném zániku jádra, klasicky v erytrocytech. Koncentrace glykogenu v erytrocytech slouží k diagnostice některých forem glykogenos (normální koncentrace pod 100mg/l lysátu isolovaných erytrocytů), hodnota nad 150 je považována za patologickou). Elektronopticky byly prokázány klasické partikule glykogenu.V erytrocytech je prokazatelná glykogen synthasa a enzymy degradující glykogen, persistující ze stadia erytroblastu. Jejich aktivita se snižuje stárnutím erytrocytu.
Podobně persistuje glykogen v krevních destičkách a v buňkách oční čočky. Zde se podle některých literárních údajů může akumulace nesolubilního glykogenu v nucleus lentis podílet svými značně konstantními fyzikálně chemickými (optickými) vlastnostmi na osově lokalisovaném refrakčním efektu např. u ptáků (dravci).
Biologický význam glykogenu: glykogen je využíván zejména jako energetická reserva. Z řady pozorování vyplývá, že je využíván i pro kompensaci některých fyzikálně chemických změn v buňce jako je pokles osmotického tlak, který může polymer glykogenu kompensovat svou velkou kapacitou vázat vodu (viz. sekundární akumulace hlykogenu). Z řady pozorování lze však usuzovat i na biologicky významnou roli glykogenu v buněčném jádře, i když bližší konkretisace není možná (viz. níže)
Histologicky je glykogen detekovatelný ve formě různě velkých cytoplasmatických granul. Detekovatelnost glykogenu v rutinních histologických řezech je problematická, pro jeho poměrně vysokou solubilitu ve vodných fixativech (formaldehyd). Určitou výhodu skýtá perfusní fixace nad fixací imersní. Při imersní fixaci dochází pravidelně a v různém rozsahu k posunu glykogenu v rámci buňky, takže jeho distribuce je změněná. Výtečně je glykogen uchován po fixaci metanolem nebo etanolem v kryostatových řezech z čerstvě zmražené tkáně (obr. 4). Velmi efektivní je imersní fixace tkáně z metanolu (případně etanolu).V patologicko anatomické praxi je detekován empirickým barvením Bestovým karmínem, nebo PAS reakcí, kontrolovanou natrávením diastasou k odlišení především diastasaresistentních glykoproteinů.
Za nejlepší metodu znázornění (tedy ne fixace) glykogenových partikulí je považována sekvence: 2%glutaraldehyd v kakodylátovém pufru, následovaná směsí 1%OsO4 + 1.5% ferrikyanid a standardním odvodněním. Za kontrast glykogenu v elektronové mikroskopii jsou zodpovědné asociované proteiny.
Akumuluje-li buňka excesivní množství partikulí glykogenu, dochází k jejich hromadění v základní cytoplasmě. To vede zpočátku k relativnímu, později i absolutnímu úbytku ostatních organel, která jsou odtlačována na periferii k buněčné membráně. Celá periferní oblast buňky je pak výrazněji barvitelná a dominuje nad vodojasnou cytoplasmou, prostoupenou glykogenem (glykogen je v rutinních barveních neznázorněn). Takto postižené buňky připomínají buňky rostlinné tkáně a je pro ně reservován termín buňky vodojasné (viz. níže). Platí to pro situace, kdy dominantou v buňce jsou partikule glykogenu; jestliže jsou současně zmnoženy i jiné organely (např. ER), pak je úměrně tomu zmíněná vodojasnost redukována.
Velmi často jsou partikule glykogenu přítomny i mimo cytosol. Nejčastěji se nalézají v jaderném kompartmentu (intranukleární inkluse glykogenu) (obr. 5), zejména u některých forem glykogenos, ale i u jiných poruch (např. dibetetes mellitus, Wilsonova nemoc) nebo u některých nádorů. Popsána byla i přítomnost glykogen synthasy v isolovaných jádrech. Velmi často byla popsána masivní deposita partikulí glykogenu intramitochondriálně (obr. 6), a to někdy i v takovém množství, že je celá vnitřní struktura mitochondrie obliterována (obr. 7).
V současnosti lze vysvětlit tyto fenomény pouze spekulativně, např. průnikem části cytosolu s komplexem syntetických enzymů a glykogeninu, případně i s preformovanými partikulemi glykogenu do jaderného nebo mitochondriálního kompartmentu (není známo, že by tyto dva kompartmenty byly postiženy současně). Nakolik by to objasnilo průkaz glykogen synthasy v isolovaných buněčných jádrech není jasné. Není rovněž známo, jak se účastní takto segregovaný glykogen na celkovém buněčném obratu glykogenu.
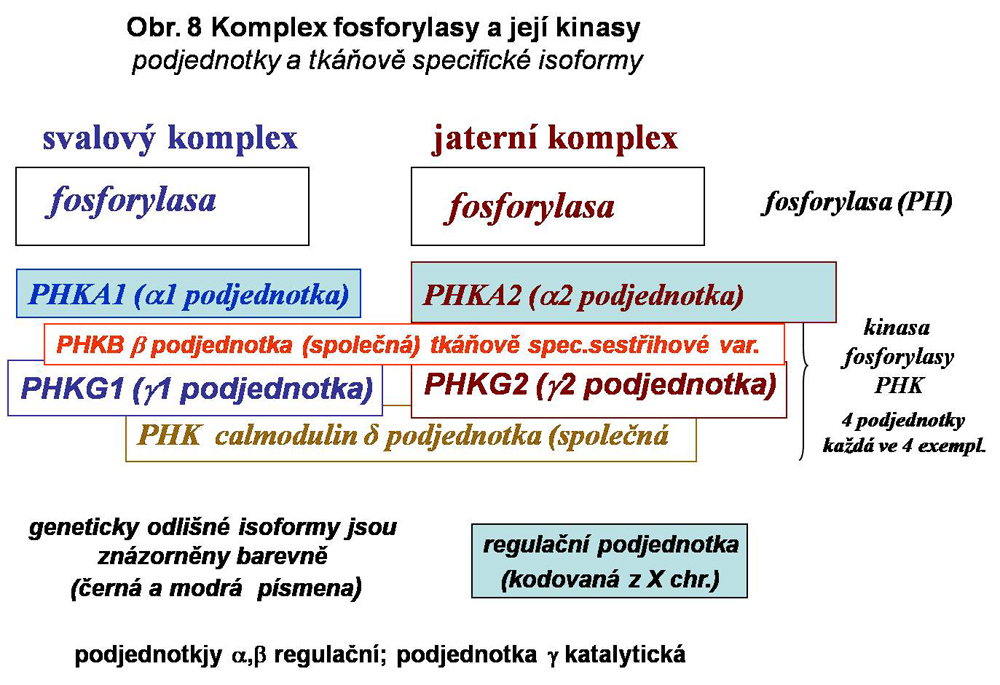
Degradace je realizována komplexem fosforylasa - odvětvující enzym situovaným v cytosolu. Fosforylasa (PH, v neaktivní formě značená jako b, v aktivní jako a) je homodimer, štěpící lineární vazbu 1-4, a to jak v hlavním řetězci, tak v řetězcích bočních. Degradace začíná na neredukujících koncích řetězců glykogenu. V bočních řetězcích je to až po poslední čtyři cukry před větvícím bodem. Fosforylasa je známa ve třech isoformách, jaterní, svalové a mozkové, kódované z odlišných genů. Stupeň jejich podobnosti je okolo 80%. Její aktivita je kontrolována fosforylací (kinasou, viz. níže; defosforylace se děje pomocí protein fosfatasy 1) a allostericky (aktivace AMP, IMP, inhibice G6P, ATP). Regulace fosforylasy je spjata s regulací glykogen synthasy. Enzymový komplex je složen z tkáňově specifických isoforem vlastního enzymu a z komplexu kinasy, která je složena ze 4 odlišných podjednotek, každá ve 4 exemplářích (heteromultimer). Některé z nich jsou tkáňově specifické nebo kódované z X chromosomu (viz. obr. 8).
Histochemicky lze znázornit aktivitu fosforylasy. V histochemické metodě je využívána syntetická aktivita fosforylasy změnou pH) (Obr. 9)
Odvětvující enzym (debrancher). Zbytek bočního řetězce, zkráceného lineárně fosforylasou je přenesen na hlavní řetězec odvětvujícím enzymem (debrancherem) - bifunkčním enzymem, který má dvě katalytická centra (amylo 1,6-glukosidása, 4- α glukantransferasa - AGL, nebo GDE - glykogen debrancher enzyme), který odštěpí triglukosid a přenese ho na konec lineárního řetězce (prvý krok aktivního centra transferásového) a v druhém kroku hydrolyzuje 1-6 vazbu (1-6 α-D glukosidasová funkce druhého aktivního centra). Tento enzym (monomer, 165kDa, 1532 aminokyselin) má řadu setřihových variant a tkáňově specifické promotory. Takto je glykogen postupně degradován střídající se aktivitou glykogenfosforylasy a odvětvujícího enzymu. Výsledkem fosforolytického štěpení (vnášení fosfátu do glykosidické vazby) je glukosa-1 fosfát, který je isomerasou přeměněn na glukosa-6 fosfát. Jeho další osud je dvojí, podle toho, zda se buňka účastní regulace hladiny krevního cukru nebo zda jde o "privátní" využití uvolněné glukosy. V buňkách neexprimujících glukosa-6 fosfatasu (viz. níže) je G6P zpracován v dané buňce zejména glykolysou pro krytí vlastní energetické spotřeby.
Existují pozorování svědčící pro to, že v situacích, vyžadujících mobilisaci glukosy z glykogenu (pro potřeby buňky samotné) nejsou degradovány glykogenové agregáty stejnoměrně. Ve svalové tkáni bylo prokázáno nezávisle několika skupinami, že glykogen je degradován při různých typech fyzické zátěže odlišně v oblasti subsarkolemální (preferenčně při sprintech) a intrasarkomerické inter- a intramyofibrilární (zde při maratonu).
Lysosomální degradace glykogenu je samostatnou kapitolou degradace glykogenu. Degradace je katalysována kyselou α-glukosidasou (enzym štěpící 1-4 i 1-6 vazby) v lysosomálním kompartmentu. Glykogen se do lysosomálního systému dostává mechanismem autofagocytosy, tedy společně s dalšími komponentami cytoplasmy. Mechanismus isolovaného přesunu glykogenu do lysosomů nebyl doposud popsán.
Podstatou akumulace glykogenu je koordinace jeho zvýšené synthesy a útlumu degradace na straně jedné s omezenou "absorpční kapacitou" stávajících agregátů glykogenu. To má za následek tvorbu nových granul glykogenu procesem zmíněným výše (viz. glykogenin). řešení situace zvětšováním granul glykogenu není tedy možné.
V následujícím textu jsou jednotlivé formy probrány podle základních patogenetických mechanismů (viz. též obr.10) než podle klasického číselného označení, které je uvedeno paralelně.
1. geneticky podmíněné primární poruchy degradace glykogenu, dané mutací enzymů angažovaných přímo v degradaci glykogenu (fosforylasa, kinasa fosforylasy, odvětvující enzym, lysosomální α glukosidasa) nebo v jeho synthese (glykogenosa typu IV a deficit glykogen synthasy).
2. nepřímé poruchy degradace glykogenu. Jde o dysregulaci obratu glykogenu ve prospěch jeho akumulace (zvýšení synthesy, spojená s inhibicí degradace). Jde o deficit G6Pasy (regulace glykémie), poruchy v glykolytickém řetězci (zpětně se promítající akumulace substrátů) a poruchu transportu glukosy. Jde o geneticky podmíněné poruchy.
3. další poruchy obratu glykogenu geneticky podmíněné a získané
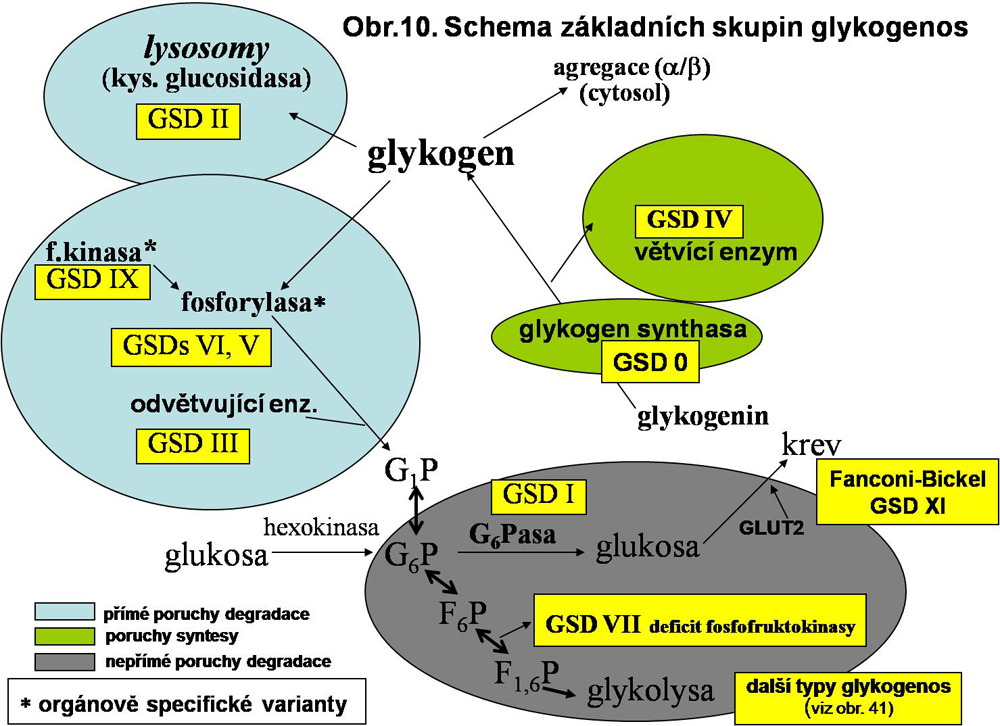
4. přiřazena je skupina tzv. amylopektinos
1. geneticky podmíněné primární poruchy degradace glykogenu, dané mutací enzymů angažovaných přímo v jeho obratu
1.1 deficit fosforylas (orgánově specifických)Všechny tyto primární genetické poruchy jsou autosomálně recesivní, s výjimkou deficitu některých fosforylaskinas (viz. obr.8)
1.1. deficit aktivity fosforylasyTyto poruchy se týkají deficitu orgánově specifických fosforylas, které jsou známé tři: jaterní svalová, a mozková (viz. výše). Toto označení je čistě formální a vychází z maximální orgánové exprese příslušné isoformy, která je vždy exprimovaná v dalších orgánech. V současnosti jsou definovány pouze deficity svalové a jaterní isoformy.
Deficit svalové fosforylasy (GSD V) je omezen na svalovou tkáň a to zejména kosterního svalu. O postižení kardiomyocytů není v literatuře referováno. Předpokládá se, že v srdečním svalu může být deficit kompensován přítomností jiných isoforem (isoformou mozkovou). Projevuje se progresivní myopatii, která může být provázena dosti výraznou destrukcí svalových vláken vystupňovanou zejména fysikální aktivitou (známky myolysy, myoglobinurie). Mechanismus interference tohoto enzymového deficitu s biologii svalového vlákna není znám. Hladina krevního cukru není poruchou ovlivněna.
Deficit jaterní fosforylasy (GSD VI) je omezen na hepatocyty a projevuje se isolovanou hepatopatii a hepatomegalii, většinou bez větších následků. Hypoglykémie nebývá přítomna. V řadě případů jsou residuální aktivity vysoké a těžko se biochemicky v těchto případech prokazuje. Histochemicky lze prokázat deficit, pokud je výrazný (obr. 11). Tento jaterní isoenzym není údajně exprimován v jiných tkáních. Biochemickou diagnosu lze tedy realizovat pouze ve vzorku jater.
O deficitu mozkové isoformy (přítomné i v jiných orgánech, např. v ledvinách, viz. níže) není doposud nic známo.
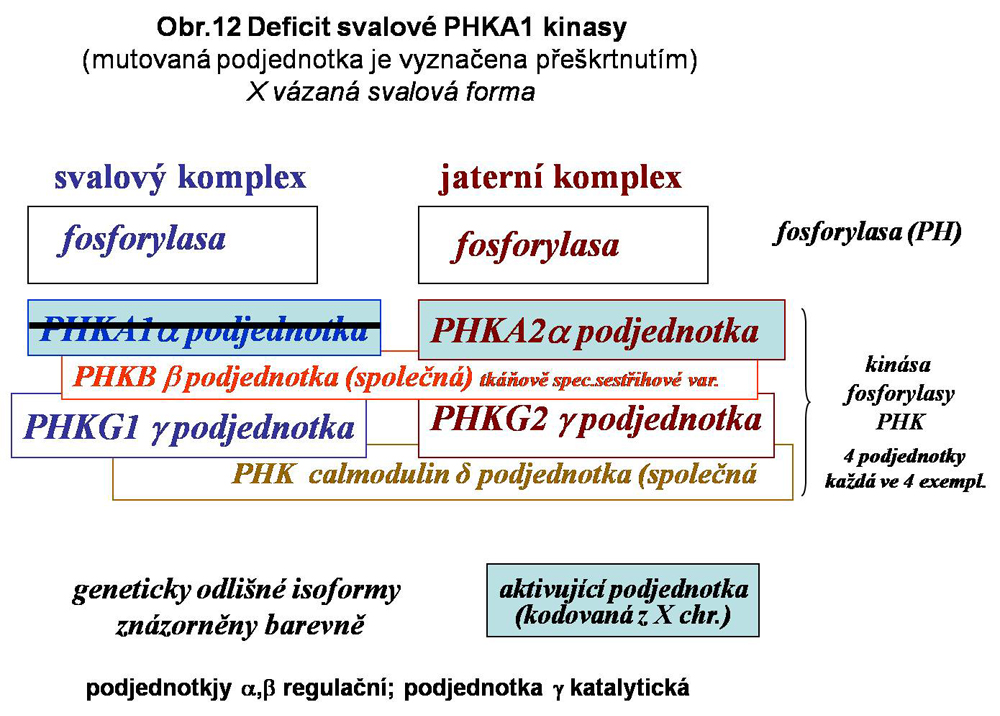
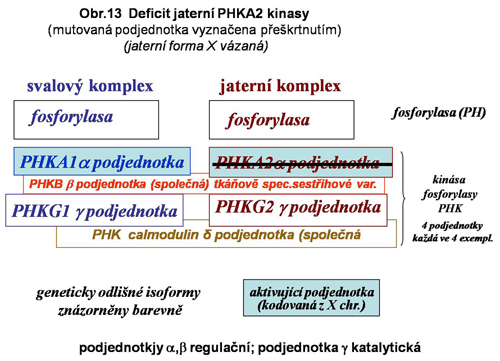
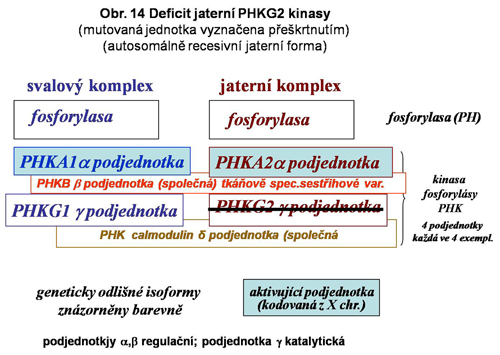
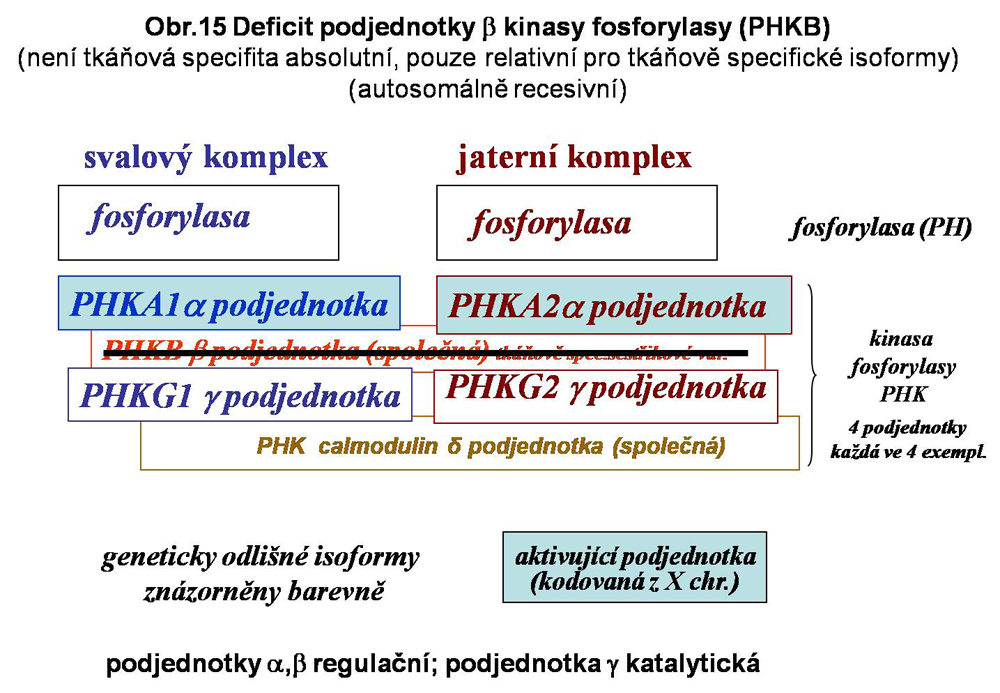
Tento heterogenní multipodjednotkový (multiheterotetramer) systém aktivace fosforylasy (změna PHb, tj.fosforylasy b = neaktivní na aktivní PHa) je do značné míry tkáňově specifický, což naznačuje existenci specifických podmínek mobilisace glukosy fosforylasou v tkáních, přičemž extrémy představuje hepatocyt a vlákno kosterního svalu (viz. výše obr.8). Přehledně jsou zmíněny doposud známé definované varianty deficitů v systému kinas fosforylasy. Situace v této kategorii poruch není zdaleka plně objasněna. Celá skupina je zařazena do stále poněkud nepřehledné a mechanické klasifikace jako GSD IX (podskupiny a,b,c a další), zčásti jako VIII. Jednotlivé spolehlivě definované varianty jsou uvedeny níže se schematy (viz. obr.12,13,14,15). Deficit kalmodulinu má pravděpodobně mnohem širší důsledky než pouhou ztrátu aktivace fosforylasy v degradaci glykogenu.
Tyto typy glykogenos se u juvenilních a adultních forem presentují jako myopatie se symptomy únavy při svalové zátěži, křečemi, často i myoglobinurii. Nejsou známky jaterního postižení a v referovaných případech není zmínka o kardiomyopatii. Ve svalových biopsiích jsou popisovány mírné akumulace glykogenu subsarkolemálně, blíže nespecifikovaná vakuolární akumulace glykogenu a některé nepříznačné degenerativní změny. Publikovány byly i časné formy infantilní s těžkým průběhem. V publikovaných případech byl deficit kinasy prokazatelný pouze ve vzorcích ze svalu. Aktivity v leukocytech a erytrocytech byly normální
a) z deficitu jaterní alfa podjednotky (PRKA2); jde o X vázanou benigní hepatomegalii; údajně jde o nejčastější formu glykogenosy (může mít zvýšené serové triglyceridy, cholesterol, zvýšené jaterní testy); popsána byla i renální tubulární acidosa.
b) z deficitu jaterní gama podjednotky (PHKG2); byla popsán i přechod do cirhosy a vznik hepatomů (není pravidlem).
 |
 |
Hepatomuskulární forma z deficitu společné podjednotky beta (PHKB); mírný průběh; autosomálně recesivní
Diagnosu deficitů kinás fosforylas lze stanovit vyšetřením aktivity enzymu v erytrocytech (s výjimkou svalových forem)
Důsledky enzymového deficitu (fosforylasy i její kinasy) pro buňku nejsou přesně definované. Obecně jde o následky snížené schopnosti mobilisace glykogenu pro metabolismus buňky samotné, pokud jde o hepatocyty tak i pro regulaci krevního cukru
Patologie těchto poruch je omezena na cytologické známky zvýšené akumulace glykogenu v hepatocytech (obr. 16) nebo v kosterním svalu.. Střádání glykogenu může být provázeno různou mírou regrese. Srdce má tendenci k hypertrofií (kardiomyopatie na podkladě glykogenosy).
Z dosavadních pozorování lze usoudit, že postižení kardiomyocytů je výjimečné. Zcela stranou je extremní případ postižení srdce u vzácné fatální formy glykogenosy lišící se od shora zmíněných orgánově tím, že postihuje kardiomyocyty. Manifestuje se časně po porodu. Byla klasifikována jako blíže nespecifikovaný deficit kinasy fosforylasy, velmi pravděpodobně specifické pro kardiomyocyty (obr. 17). Za zmínku stojí, že v některých infantilních fatálních kardiálních glykogenosách byl prokázána dysfunkce AMP aktivované protein minusy (mutace v její podjednotce gamma 2)
Skupina deficitu PH/PHK není stále považována za plně objasněnou na molekulární úrovni.
Degradace glykogenu je omezena na zevní řetězce, které efektivně zkracuje fosforylasa až po prvé větvení, takže uvolňování glukosy pro energetické potřeby buňky ev. pro regulaci hladiny krevního cukru je silně omezena. Přebytek glukosy z potravy a z glukoneogenese je řešen silně omezeným doplňováním zevních řetězců glykogenu, zejména však zvýšenou synthesou glykogenu, který se zákonitě hromadí. Buněčný glykogen má tendenci ke kratším zevním řetězcům a tedy ke zvýšené solubilitu (i když dokonalé vysvětlení zvýšené solubility neexistuje). Z uvedeného není jasné, čím tato porucha interferuje významněji s biologii postižených buněk. Manifestace je v jaterní tkáni (hepatopatie a hepatomegalii, tendence k fibrose) a v tkáni svalové (myopatie), včetně myokardu (kardiomyopatie), i když stupněm postižení se mohou tkáně od sebe dosti výrazně lišit. Stupeň postižení však není dramatický a průběh bývá velmi protrahovaný. Velmi často jsou výrazné známky hepatopatie v dětství, které však z větší části časem ustupují.
Histologický projev je dán akumulací glykogenu. V játrech jsou prokazatelná i glykogenová jádra, mírná steatosa a tendence k fibrose (obr. 18) (obr. 19), vzácně k cirhose. Bývá hyperlipoproteinemie. Mechanismy, vedoucí k poškození buňky a k dalším biochemickým projevům nejsou známé.
Existuje dělení na GSD IIIa (nejčastější jaterní a svalová forma, časté je postižení srdce), méně častá forma IIIb (jaterní). Udávají se výjimečné formy IIIc (isolovaná svalová forma, se selektivním postižením transferásové aktivity) a IIId se selektivním postižením glukosidasové aktivity).
Diagnosa se opírá o zvýšenou koncentraci glykogenu v erytrocytech (viz. výše), stanovení deficitní enzymové aktivity v bioptických vzorcích z tkání,v tkáňové kultuře fibroblastů, v erytrocytech. Průkaz mutací v příslušném genu může narážet na existenci sestřihových variant (viz. výše).
Jde o deficit lysosomální α glukosidasy, podmiňující deficit štěpení glykogenu v lysosomech (obr. 20). Jde o prvou lysosomální střádací poruchu u níž byl popsán enzymový deficit. V lysosomech se glykogen hromadí ve formě beta granulí. Jelikož není znám transport glykogenu do lysosomálního systému lze předpokládat, že přísun glykogenu do tohoto kompartmentu je realisován obecným mechanismem autofagie. Intenzita autofagie a množství cytosolického glykogenu by tedy měly představovat hlavní faktory určující intensitu poruchy v dané buňce. S tím souhlasí to, že častým projevem na úrovni buňky jsou fenomény autofagie s řadou organel sekvestrovaných do lysosomálního systému, který v konečném stadiu obsahuje prakticky čistou populaci granul glykogenu, neboť ostatní sekvestrované části cytoplasmy byly úspěšně degradovány. V časných formách (infantilní varianta) má porucha tendenci ke generalisaci s maximem v kosterním, srdečním svalu, hepatocytech (obr. 21) (obr. 22) (obr. 23) (obr. 24), ale i v hladkém svalu, s postižením i CNS s maximem v míšních neuronech (obr. 25) Výrazné postižení lysosomální střádáním bylo popsáno i v sítnici, včetně neuronů. Po stránce klinické dominuje střádací hepatopatie s hepatomegalii, kardiomyopatie (hypertrofická) a myopatie. Je pozoruhodné jak kvantitativně rozličné postižení mohou vykazovat vlákna v jednom vzorku kosterního svalu. Masivní postižení míšních α motoneuronů je pravděpodobně zodpovědné za výraznou hypotonii.V pomaleji probíhajících formách se postižení koncentruje na svalový aparát, který je u dospělé formy prakticky výlučně postižen (myopatická forma) (obr. 26) (obr. 27)
V bioptických vzorcích byl měly být projevy lysosomálního střádání. V cytologickém obraze pak dominuje vakuolisace s parametry lysosomálního systému, t.j. zvýšená aktivita kyselé fosfatasy v kryostatových řezech, přítomnost katepsinu D a případně membránových markerů lysosomálního systému (viz. výše). Pokud je glykogen zachován (je redukován imersní fixací i autolysou) je prokazatelný běžným barvením. V elektronovém mikroskopu je prokazatelný intralysosomální glykogen a projevy autofagocytosy. Výrazně zmnožen je i extralysosomální glykogen. Poslední výzkumy naznačují, že ve svalové tkáni je zatím ne zcela objasněným mechanismem vystupňovaná aktivita autofagocytosy, která je navíc ne zcela efektivní, což vede k dalšímu poškození buněk. Efektivita terapie podávání rekombinantního enzymu je v tomto ,,komplikovaném" terénu minimálně (efektivní) přínosná.
Jako jediný korelát pro klinickou biochemii na úrovni metabolitu je častý, nikoliv však konstantní průkaz degradačního produktu glykogenu ve formě tetrasacharidu (tetraglukosid) v plasmě a v moči. Není však považován za zcela specifický pro GSD II. Nicméně je všeobecně používán pro screening poruchy a to i pro adultní formy.
Biochemicky je prokazatelný deficit kyselé alfa glukosidasy s různou residuální aktivitou. Biochemická diagnostika je ztížená přítomností neutrálních glukosidas, které mohou v různé míře interferovat, zejména u pomaleji probíhajících forem s vyšší residuální aktivitou. Tato interference je značná ve vzorcích z periferních leukocytů, běžně užívaných pro vyšetření. Zavedení nové metody stanovení aktivity kyselé alfa glukosidasy v přítomnosti akarbosy, která inhibuje neutrální glukosidasy, umožňuje pro diagnostiku použít i leukocyty, event. suché kapky krve (pro screening). U dospělých forem s různě vysokou residuální aktivitou kyselé alfa glukosidasy se přesto doporučuje prokazovat enzymový deficit rovněž v kultivovaných kožních fibroblastech, kde jsou neutrální glukosidasy minimálně exprimované.
Molekulárně genetická analýza. Biochemická diagnosa je zpravidla doplněná analysou mutací v genu α glukosidasy
Dříve popisovaná GSD II bez enzymového deficitu (zvaná GSD IIb, Danonova nemoc) byla vysvětlena jako porucha, způsobená mutací integrálního glykoproteinu lysosomálních membrán LAMP2. O této je blíže pojednáno v oddíle lysosomální systém a jeho poruchy. Vyznačuje se procesem připomínajícím excesivní deregulovanou autofagocytosu s přítomností glykogenu v lysosomech, což do určité míry připomíná buněčnou patologii Pompeho nemoci
Tato porucha není glykogenosou v pravém slova smyslu, protože výsledná situace je zrcadlovým obrazem glykogenosy neboť v postižených tkáních je snížené množství až absence glykogenu. Existují dvě formy, dané mutací v isoenzymech GS.
Jaterní forma (deficit GS 2) je doposud nejlépe známá, i přes velmi malý počet diagnostikovaných případů. Vzhledem k tomu, že je jaterní enzym exprimován pouze v játrech, symptomatologie je daná dysfunkcí regulace hladiny krevního cukru. Základním narušeným procesem je nemožnost inkorporovat glukosu do jaterního glykogenu. Glukoneogenesa je neporušená, rovněž β oxidace mastných kyselin (která je kompensačně zvýšená). Příjem glukosy (potravou nebo při zátěžovém testu) má za následek persistující hyperglykemii s ketonurií (situace připomíná časný diabetes mellitus). Příčina postprandiální hyperglykémie není zcela jasná; je však pravděpodobně ve vztahu k deficitní synthese glykogenu. Při hladovění je tendence k hypoglykemii při vysoké ketonurií. Ketonurie (kompensačně zvýšená β oxidace) je do značné míry ochranným mechanismem neuronů před atakami hypoglykémie. Prognosa je variabilní, jsou známé i protrahované případy. Diagnosa se opírá zejména o průkaz mutací v GS 2 genu po podrobném klinicko biochemickém vyšetření. Jaterní biopsie je rovněž zvažována, ale výsledky nemusí být údajně jednoznačné. V biopsiích byl glykogen redukován, ale částečně (v malém množství) persistoval (podíl GS 1?). Může být výrazná steatosa hepatocytů. Obecně lze říci, že nedochází k výraznějšímu poškození jater.
Deficit svalové formy enzymu (GS 1), exprimovaného ve svalech, mozku a v ledvinách je známo velmi málo. V doposud ojedinělých případech šlo o manifestaci myopatickou a kardiomyopatickou (hypertrofická KMP) v dětském věku. V biopsiích je deficitní glykogen, zmnožené mitochondrie, hypertrofie kardiomyocytů. Jaterní glykogen je v normálním množství.
Lze předpokládat, že v úvahu může přijít vedle mutace GS i mutace v glykogeninu (jaterním nebo svalovém, viz. výše), který by se mohl projevovat způsobem velmi blízkým deficitu glykogen synthasy
Deficit tohoto enzymu (GBE-glycogen branching enzyme) má za následek tvorbu převážně lineárního glykogenu glykogen synthasou, což není provázeno dostatečným větvením a tím nemožnost agregace do typických granul (viz. výše). Syntetizován je glykogen abnormální, nedostatečně větvený (kvalitativní porucha). Tím připomíná amylopektin, který je však chemiky definován jako součást rostlinné buňky. Je to jediná glykogenosa na basi poruchy synthesy. Jejím významným projevem na úrovni buňky je tvorba tělísek, nazývaných polyglukanová nebo amylopektinová nebo historickým názvem corpora amylacea (škrobová zrníčka), která obsahují fibrilární struktury. Blíže je o těchto tělíscích pojednáno níže, protože nerozlišitelná tělíska se vyskytují u několika dalších stavů.
Fenotypové spektrum GSD IV je neobyčejně široké a zahrnuje formy kongenitální, neonatální, včetně hydrops fetus, časné postnatální formy infantilní, protrahovanější formy juvenilní až adultní. Mezi postižené orgány patří játra, kosterní sval, myokard, CNS, méně další (např. leukocyty, epidermis, potní žlázky). Z dosavadních pozorování dále vyplývá, že není úměra mezi mírou residuální aktivity a tíží průběhu. Dále skutečnost, že enzymový deficit nemusí být prokazatelný mimo postižené tkáně (např. v leukocytech nebo kultivovaných fibroblastech při postižení kosterního svalu nebo myokardu !), ale dokonce i v tkáni postižené. Pozoruhodná je i obecná diskrepance v postižení výše zmíněných tkání. Pouze u klasické infantilní varianty je progresivní hepatopatie, myopatie a kardiomyopatie. Popsány jsou varianty s dominantně postiženým myokardem (i adultní), s dominantně postiženými játry (progresivní forma, ale i pozoruhodně stacionární forma). Extremní variantou je dospělá forma, zvaná též APGBD (adult polyglukosan body disease) s periferní neuropatii (polyglukosanová tělíska jsou v periferních nervech), myopatii a případně celým spektrem neurologických příznaků (axonální akumulace tělísek v CNS).
U těžkých infantilních generalizovaných forem je vznik tělísek v CNS omezený převážně na astrocyty, ale v některých případech byl popsán i intraneuronální projev střádání, zejména v míšních neuronech (viz. též níže). Pokud nejsou postižena perikarya neuronů, mohou být tělíska přítomna v neuronálních výběžcích.
Výrazná přítomnost polyglukosanu by měla mít za následek tendenci k hypoglykemiím, protože účinnost fosforolytického štěpení glykogenu je úměrné množství větvení (viz. úvod). O charakteru glykogenu v erytrocytech není známo nic bližšího. O zmnožení glykogenu blízkého amylopektinu v erytrocytech jsou literární údaje rudimentární.
V diferenciální diagnose APGBD přichází v úvahu identický fenotyp bez enzymového deficitu (viz. též níže). Na překážku spolehlivého hodnocení některých diskrepancí ve skupině GSD IV (viz. výše) je kvalita v současnosti dostupných biochemických metod, které jsou nepřímé a neumožňují citlivé kvantitativní hodnocení residuální aktivity. Vždy by měla být pro dokonalé hodnocení realisována i analýza příslušného GBE genu.
Za zmínku stojí i skutečnost, že oproti ostatním glykogenosám postihujícím difusně cytosol (s výjimkou GSD II, viz. obr. 42) je deposice polyglukanových tělísek v cytoplasmě u GSD IV fokální a že jsou v postižené buňce velmi často pozorovaná i normální granula glykogenu jak o α tak o β struktuře. To svědčí o částečně zachované synthese glykogenu. Pravděpodobně je to dáno residuální aktivitou enzymu. Bohužel, u publikovaných případů s nulovou aktivitou větvícího enzymu nebyla přítomnost normálních granul glykogenu systematicky sledována.
Výlučné postižení pouze části populace granul glykogenu by mohla tak svědčit o jejich biologické heterogenitě (viz. výše glykosomy).
2. nepřímé poruchy degradace glykogenu způsobené deregulací obratu glykogenu ve prospěch jeho akumulace (zvýšení synthesy, spojená s inhibicí degradace).
2.1 deficit glukosa-6 fosfatasy - enzymového systému regulujícího hladinu krevního cukru
2.2 deficity glykolytického řetězce
2.3 defekt transportéru glukosy (GLUT 2)
2.1 deficit aktivity glukosa-6 fosfatasy (GSD I A, GSD I nonA
(podle enzymové nomenklatury je správný název enzymu glukosa-6-fosfát fosfatasa)
Komplex glukosa-6 fosfatasy. V buňkách exprimujících aktivitu glukosa-6 fosfatásy (viz. níže) dojde k uvolnění glukosy, která je transportována přes buněčnou membránu (GLUT2 transportér) do extracelulárního prostoru a doplňuje tak hladinu krevního cukru (obr.28).
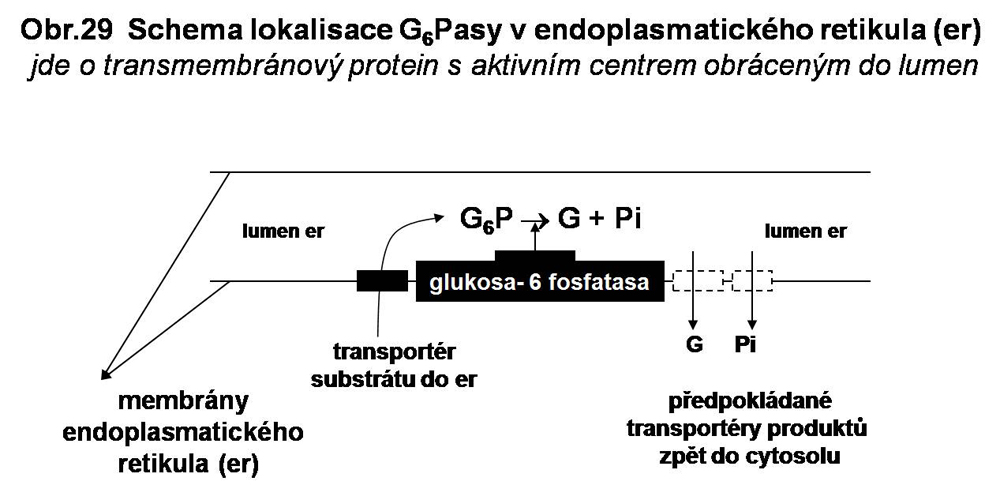
Komplex glukosa-6 fosfatasy je lokalisován v membránách endoplasmatického retikula (včetně perinukleární cisterny). Skládá se z glukosa-6 fostatasy samotné (transmembránový protein, 35 kDa, 357 aminokyselin) s aktivním centrem v lumen endoplasmatického retikula, funkčně napojený na transportér zodpovědný za přísun G6P substrátu do endoplasmatického retikula. Transportér G6P je transmembránový protein o 429 aminokyselinách. Je ubikvitní a je velmi pravděpodobně je napojen na všechny existující G6Pasy (viz. níže). Stále se spekuluje o existenci dalších transportérů, které by byly zodpovědné za transport produktů (glukosy a Pi) zpět do cytosolu (viz. obr 29). Z recentních studiích studií vyplývá, že G6P transportér vykazuje současně i vlastnosti transportéru Pi (Pí antiporter).
V současnému datu jsou známy tři isoformy G6Pasy), které jsou exprimovány v různých buněčných typech. To vysvětluje přítomnost histochemicky prokazatelné enzymové aktivity v celé řadě buněčných typů (např. lidský trofoblast, a další, studované zejména v tkáních laboratorních zvířat). G6Pasa byla detekována v endoplasmatickém retikulu i v makrofázích, což pomohlo definovat účast membrán ER na tvorbě membrány fagosomu (mimo to i jiných buňkách), kde pomohla prokázat podobnou účast ER na vzniku autofagosomu
Ve skupině glykogenos je to isoforma α (αGPasa, zvaná též C1), jejíž mutace je zodpovědná za GSD IA. Jsou známy tři buněčné typy exprimující tuto αG6Pasu - hepatocyt, ve kterém je situace nejlépe probádaná, dále epitel proximálního tubulu ledvin a enterocyt. Regulace hladiny krevního cukru je spojována nejvíce s hepatocyty a s renálními tubuly. Cytosolový glykogen je degradován klasickou cestou, uvolněná glukosa (resp. glukosa-6-fosfát) je defosforylována a transportována na basalní pól a zde předávána do krve. O roli enterocytů v tomto smyslu není v podstatě nic známo. V hepatocytech je enzym lokalizován v ER bez predilekcí. V ledvinách a v enterocytech je jeho lokalisace omezena na ER převážně v supranukleární části (viz. obr. 30).
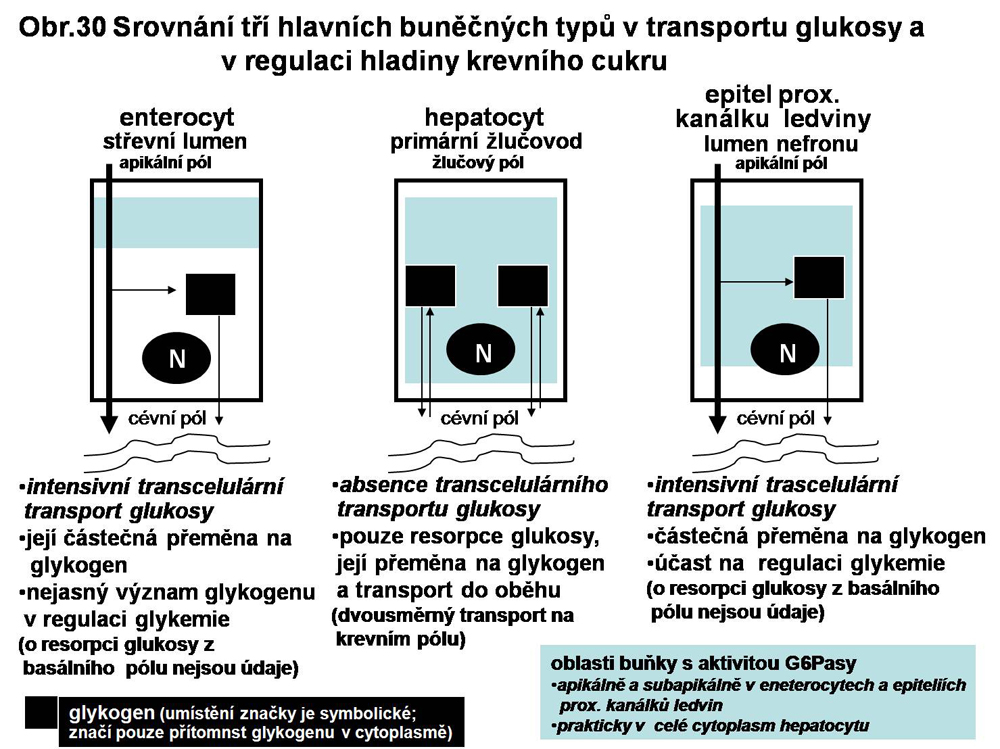
Glykogen se u GSD IA pravidelně akumuluje (viz. níže) v hepatocytech a v epitelu proximálního tubulu. Nic známého není ani o střádání glykogenu v enterocytech u GSD IA a o případných funkčních následcích.
αGPasa (zvaná též C1) je induktivní enzym, jehož exprese je realisována na úrovni transkripce pomocí SRC-2 (steroid receptor coactivator 2). Vyřazení tohoto koaktivátoru v experimentu navozuje stav velmi blízký deficitu GSD IA.
Druhá isoforma G6Pasy , zvaná C2, je exprimovaná pouze v β buňkách Langerhansových ostrůvků pankreatu. Její význam není doposud objasněn.
Třetí G6Pasa, (zvanáC , některými též β) je ubikvitní. Její vyřazení v experimentu vede pouze k mírným příznakům
Histochemicky lze aktivitu G6Pasy znázornit Gomoriho metodou (obr. 36)
2.1. GSD IA (Gierkeho hepatorenální glykogenosa) je způsobena deficitem glukosa-6 fosfatasy samotné.
Tento enzym je lokalisován v endoplasmatickém retikulu hepatocytů, epitelií proximálních kanálků ledvin a enterocytů a je klíčovým enzymem, který uvolňuje glukosu z glukosa-6 fosfátu (G6P) a tím její přenos do krve. Neštěpený G6P, který nemůže být z buňky transportován a hromadí se v deficitních buňkách. Tím nastává tendence k těžkým hypoglykemiím. Akumulace G6P v buňkách vede k defosforylaci (inaktivaci) glykogenfosforylasy na straně jedné a k defosforylaci (aktivaci) glykogen syntasy a současně její allosterické aktivaci (viz. výše). To vše přispívá k positivní bilanci glykogenu.
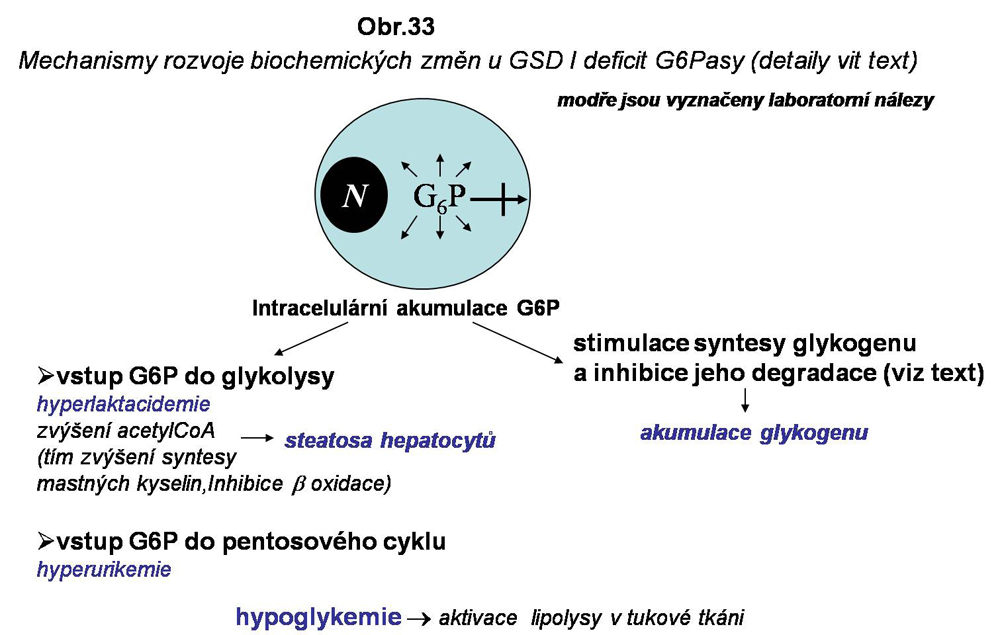
V hepatocytech porucha dominuje. Hepatocyty jsou přeplněné glykogenem, vodojasné, organely jsou vytlačeny na periferií buňky (obr. 31), pravidelně je steatosa (obr. 32). Abnormálně akumulovaný neštěpený G6P musí být metabolicky alternativně zpracován (viz. obr.33). Tímto alternativním abnormálním zpracováním se dají vysvětlit nálezy v séru: hyperlaktacideme je vysvětlitelná zvýšeným vstupem do glykolysy. Zvýšená glykolysa vede k zvýšené produkci acetyl-CoA v mitochondriích a přes citrátový cyklus k přenosu acetátu do cytosolu (detaily viz. učebnice biochemie), kde je zpětně konvertován na acetyl-CoA, za aktivace acetyl-CoA karboxylasy (produkce malonyl-CoA), kterou je zahájen proces synthesy mastných kyselin. Malonyl-CoA je zároveň velmi efektivní inhibitorem vstupu mastných kyselin do mitochondrie (inhibice CPT I - vit kapitolu steatosy z poruch β oxidace. Tak lze vysvětlit steatosu, která je průvodním jevem při akumulaci glykogenu u GSD I. Hyperurikemie je vysvětlována zvýšeným vstupem G6P do pentosového cyklu.
Mechanimus časté hyperlipidemie není příliš jasný. Zvýšená synthesa VLDL nebyla prokázána. K hyperlipemii údajně přispívá snížená přeměna VLDL na LDL v periferii a snížená klírens sérových lipidů)
Na úrovni histologické se tedy deficit G6Pasy projevuje v jaterních buňkách abnormální světlostí (akumulace glykogenu) a vakuolisací (steatosa). V některých případech může být steatosa hepatocytů dominantním projevem (obr. 34) (obr. 35).
Renální postižení u GSD I je charakterizováno akumulací glykogenu v buňkách proximálního kanálku ledvin (všech jeho úseků, tedy pars convoluta a pars recta) a k nefromegalii. To může vést k poruše resorpční kapacity v této části nefronu a k rozvoji sekundárního Fanconiho syndromu mnohočetných tubulárních defektů (aminoniacidurie, glukosurie, hyperfosfaturie). Zároveň je známo, že dochází ke glomerulární hyperfiltraci a progresivnímu poškození glomerulů s rozvojem proteinurie a glomerulosklerosy.
Srovnávací studie s diabetem, a se stavy charakterisovanými glukosurii ukázaly, že glykogen se za těchto stavů střádá v pars recta proximálních kanálků (tzv. Armaniho zona).
Co do postižení enterocytů v rámci GSD I je známo velmi málo (viz. výše). Ojedinělé práce popisují absenci enzymu v enterocytech studovaných v enterálních biopsiích, ale minimální zvýšení obsahu glykogenu, který je za normální situace lokalizovaným v malém množství převážně v basální části enterocytů. U GSD IA(in IB) zmnožení bylo velmi malé a nevedlo k obrazu srovnatelnému s hepatocyty a renálními tubuly. Při hodnocení specifické aktivity G6Pasy je nutno brát v úvahu aktivitu alkalické fosfatasy, která G6P také štěpí.
Varianty fenotypu. Převažuje infantilní forma. Známy jsou i mírněji exprimované juvenilní a adultní formy. Léčba je symptomatická. Velmi důležité je ovlivnění dietou. Množství cukru by mělo být upraveno tak, aby nedocházelo k jeho nadbytečnému přísunu, který by vedl k syntese glykogenu, který nelze degradovat a pokud k degradaci dojde, musí být vzniklý G6P abnormálně zpracován.
V dlouhodobé perspektivě vede GSD I k rozvoji nádorové transformace hepatocytů (hepatomy, hepatocelulární karcinomy) a k nefrosklerose (glomerulosklerose). Přítomnost více isoforem může hrát roli v kompensaci příznaků u deficit jedné z nich, klasicky u GSD IA (deficit C1 isoformy)
Diagnostika GSD I. Exprese enzymu je pouze ve třech zmíněných buněčných typech, takže respektováním neinvasivního způsobu diagnosy není dostupná tkáň pro hodnocení aktivity G6Pasy. Pokud by při podezření na GSD I k hodnocení změn v játrech byla přece jen biopsie zvolena, měla by být tkáň zpracována komplexně s použitím histochemické techniky pro stanovení aktivity enzymu (obr. 36). V běžné praxi je po zhodnocení klinických nálezů a nálezů klinické biochemie přistupováno přímo k analyse genu, ve kterém bylo dodnes identifikováno velké množství různých mutací
GSD I nonA. Její molekulárně biologickou podstatou je nedostatečné štěpení G6P při aktivní glukosa-6 fosfatase. Příčinou je selhání transportních mechanismů zprostředkujících přísun G6P do endoplasmatického retikula k enzymu. Mutací transportéru G6P (viz. výše) do endoplasmatického retikula. Deficity dalších transportérů, přenášejících produkty z endoplasmatického retikula zpět do cytosolu (viz. obr.6) nebyly doposud spolehlivě prokázány. Pokud má být diagnosa stanovena biochemicky, musí být aktivita měřena v intaktní tkáni jater, což umožňuje posouzení efektivity transportních mechanismů, dopravujících substrát k enzymovému proteinu v endoplasmatickém retikulu. Pokud by byla narušena integrita vzorku jaterní tkáně, došlo by k přímému kontaktu substrátu s enzymem samotným a výsledek by neodrážel situaci in vivo. Z toho plyne, že histochemické hodnocení aktivity v kryostatových řezech je zcela neefektivní, neboť ukáže normální aktivitu (obr. 37).
Celkový fenotyp na klinicko patologické úrovni (obr. 38), včetně sérových nálezů je takřka identický s typem IA. Studií hepatocytů je velmi málo, ale ukazuje se, že se vedle akumulace glykogenu a steatosy rozvíjí lysosomální aktivace ve smyslu autofagocytosy a mírné abnormality mitochondrií (obr. 39) (obr. 40). Významným rozdílem a zároveň rysem GSD IB je velmi častá neutropenie, jejíž mechanismus není doposud spolehlivě vysvětlen. Může být i porucha myeloidní diferenciace. Ukazuje se, že významnou roli může hrát stres endoplasmatického retikula vedoucí k apoptose leukocytů. Závažným důsledkem je imunologický deficit, vedoucí k četným závažnými infekčním komplikacím (např.záněty GIT)
Diagnosa. Biochemické vyšetření nativní jaterní tkáně (viz. výše). Konečná diagnosa je na úrovni DNA.
Poznámka. V molekulární patologií GSD IB (asi vhodněji GSD I nonA) je asi reálné uvažovat o mnohočetném deficitu G6Pás (tedy všech doposud tří známých), protože transportér G6P je ubiukvitní a je prokazatelně asociován funkčně zatím se dvěma ze tří G6Pás (C1 a C3).
Porucha v glykolytickém řetězci vede ke zvýšení koncentrace substrátu deficitního enzymu, zpětně inhibující předcházející enzymové aktivity. Za rozhodující se pokládá zvýšení koncentrace G6P, který z důvodů podobných jako u GSD I (viz. výše) vede k akumulaci glykogenu. Čím výše v glykolytickém řetězci se porucha nachází tím je tendence k akumulaci glykogenu větší. Tady je situace zvláštní tím, že byla opakovaně popsána deposice polysacharidu amylopektinového typu pravděpodobně z asynchronie mezi stimulovanou GS (lineární synthesa) a méně aktivním větvícím enzymem (viz. níže corpora amylacea). Tato deposita jsou PAS positivní, diastasa resistentní. Takováto deposita však nebyla nikdy popsána u deficitu G6Pasy. Přehledně jsou tyto stavy podány na obr.41. Zmíněny jsou v omezeném množství.
Obr. 41 GSD způsobené poruchou glykolytického řetězce
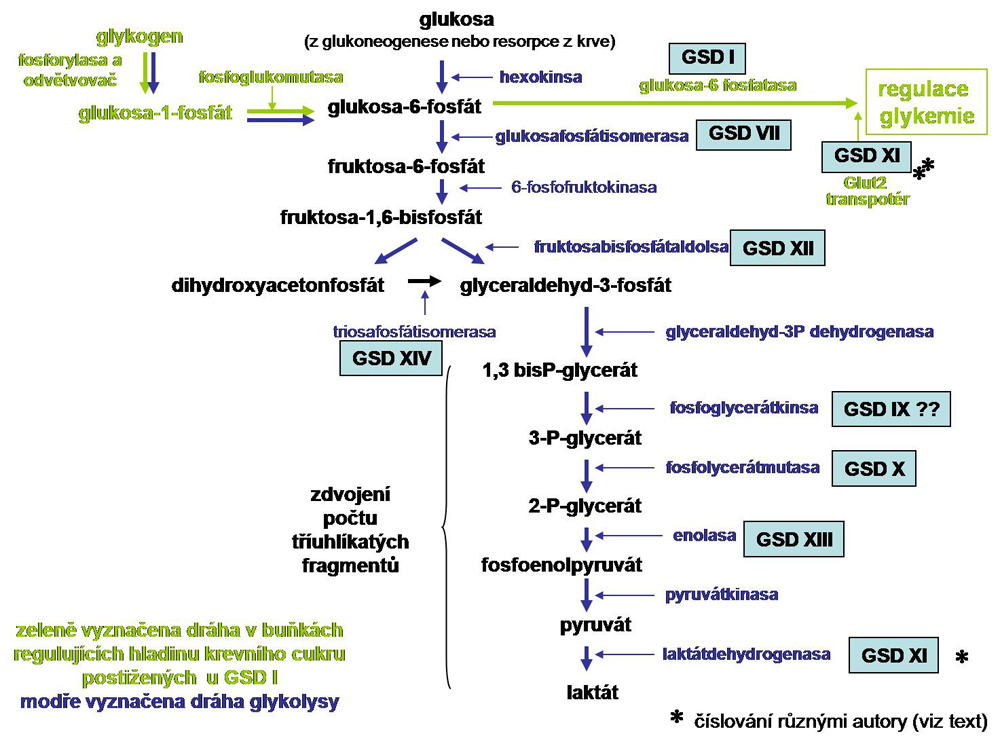
Enzym katalysující přeměnu fruktosa-6-fosfát na fruktosa-1,6-difosfát (klíčový regulační enzym glykolysy) je tetrametr, který se skládá z různě zastoupených podjednotek (M svalová, L jaterní, P destičkové). V játrech jde o homotetramer L, ve svalech homotetramer M, v erytrocytech jde o různý poměr L a M. Obecně však platí, že poměry podjednotek se mohou lišit. Tento enzymový deficit se projevuje jako různě vyjádřená myopatie. Známy jsou dětské těžké formy a mírnější formy adultní s tendencí k rhabdomyolyse. Častá bývá hemolysa, v různé kombinaci s myopatickými změnami a hyperurikemií (včetně dny). To je významné pro diferenciální diagnosu proti GSD V (deficit svalové fosforylasy, viz. výše), které nemá známky mimo svalového postižení.
Za příčinu hemolysy je považován deficit ATP z nedostatečné glykolýzy, která je hlavním zdroj energie erytrocytu. Hyperurikemie (z degradace AMP) je klasifikována jako "myogenní", vznikající v závislosti na metabolické krisi v zatěžovaném svalu, podobně jako u jiných glykogenos postihujících sval, ale ve větší míře díky postižení glykolysy . O postižení jater není nic podstatného známo.
Jde o svalovou glykogenosu danou defektem v terminální části glykolytického řetězce. Projevuje se zejména rhabdomyolysou indukovanou námahou. V biopsií jsou považované za příznačné tzv. tubulární agregáty v subsarkolemální posici a zmnožení glykogenu. Povaha těchto agregátů není jasná.
Molekulární podstata poruchy tkví v mutaci jednoho z transportérů glukosy (Glu-2), který je exprimován mimo jiné, významně v hepatocytech, enterocytech, v renálních proximálních tubulárních epiteliích a v β buňkách pankreatických ostrůvků. Narušení jeho funkce je vysvětlováno jako neschopnost uvolnit glukosu a galaktosu (fruktosa se vstřebává výhradně po po koncentračním spádu usnadněnou difusí využitím facilitujícího transportéru Glu-5) do oběhu na basolaterálních pólech zmíněných buněčných typů, ale zčásti i jejich vstup do buněk. Funkčně to má za následek kombinaci hyporesorpce zmíněných monosacharidů z GIT (kombinace malabsorpce), i když jejich postprandiální vzestup je možný, se sníženou utilisací játry, sníženou citlivostí β buněk pankreatu na hladinu glukosy (a tím sníženou sekreci insulinu) a se sníženou resorpcí ledvinami Takto zadržovaná glukosa indukuje zvýšenou synthesu glykogenu a zároveň jeho sníženou degradaci, což vede k obrazu hepatorenální a enterocytární glykogenosy do značné míry připomínající hepatorenální glykogenosu z deficitu G6Pasy (viz. výše), včetně hyperlipoproteinemie. Navíc jsou pravidelně vyjádřené poruchy resorpce v oblasti proximálního kanálku odpovídající tzv. Fanconiho syndromu se všemi důsledky (vitamin D resistentní křivice, poruchy růstu, a další).
Jaterní biopsie ukáže změny ve smyslu glykogenosy, ale bez enzymového deficitu fosforylasy , odvětvujícího enzymu a G6Pasy. V moči jsou známky komplexní poruchy resorpce: glykosurie, galaktosurie, aminoacidurie, hyperfosfaturie). Konečná diagnosa - průkaz mutací v GLUT-2 genu.
Geneticky podmíněné poruchy (žádná z nich nemá standardní číselné označení)
Mutace AMPK (proteinová kinasa aktivovaná AMP) vedoucí k neregulované aktivitě této kinásy, což má za následek aktivaci celé řady procesů, včetně zvýšené resorpce glukosy aktivovaným GLUT-4 transporterem. V dosavadních pozorováních byla mutace prokázána v regulační podjednotce gama (typ 2). To má za následek indukci zvýšení synthesy glykogenu, v některých případech bez příslušné aktivace větvícího enzymu (viz. níže).
U poruchy projevující se v dospělosti je běžně popisováno pouze postižení srdce, vzácně je zmínka o manifestaci v kosterním svalu, která je však velmi mírná (manifestuje se pouze mírným zmnožením normálních mitochondrií). Pozoruhodné je však pozorování poukazující na význam mutace PRKAG2 v infantilní fatální formě glykogenosy (viz. výše)
Z genetických poruch sem dále patří i Laforova epilepsie, podrobně popsaná níže. Excesivní akumulace glykogenu byla popsána i u některých defektů cyklu močoviny (obr. 42) (obr. 43), méně u jiných definovaných metabolických poruch (obr. 44)
Jde o stavy, za kterých se hromadí granula glykogenu v množství jako v rámci některých geneticky podmíněných glykogenos. Jde o projevy omezené na určitý buněčný typ
Výrazná akumulace glykogenu v jaterních buňkách byla popsána zejména jako následek hepatitidy. Takováto porucha, jejíž příčina není přesně známa, může persistovat delší dobu po odeznění zánětu a dosáhnout intensity srovnatelné s geneticky podmíněnou glykogenosou (obr. 45) (obr. 46). Za příčinu lze považovat narušení funkce endoplasmatického retikula (aktivity G6Pasy).
Výrazná difusní akumulace glykogenu v hepatocytech byla popsána u diabetu mellitu typu I, u hemodialysovaných pacientů, při imunosupresi po transplantaci a u některých dalších stavů (zánětlivá střevní onemocnění). Může připomínat do jisté míry tzv. matnicové hepatocyty u virové hepatitidy typu B.
Známá jsou tělíska v hepatocytech, složená z hyperplastického hladkého endoplasmatického retikula (SER) s liniemi glykogenových roset ("SER-glycogen" corpuscles).
Byla popsána dokonce i tělísková forma, připomínající do značné míry Laforova tělíska (viz. níže), opět jako reaktivní (sekundární) změna při terapii závažných onemocnění.
Poznámka - cytoplasma hepatocytů i přes výrazné zmnožení granul glykogenu nemusí být vodojasná, ale kompaktní (matnicová), čímž se odlišuje od situace u glykogenos. Vysvětlením tohoto rozdílu může být jiný typ eosinu použitého v barvení. Jiným vysvětlením je zmnožení ostatních organel v oblasti akumulace glykogenu za těchto stavů.
K výrazné akumulaci glykogenu u diabetu dochází často v hepatocytech jako následek pasivního průniku glukosy (nezávisle na insulinu) u rozkolísaného diabetu při hyperglykemií
V kanálcích ledvin může dojít k masivní akumulaci glykogenu jako následek prolongované hyperglykemie a glykosurie. Nejpostiženějším úsekem jsou partes rectae na hranici kory a dřeně (pyramid) - zona střádání glykogenu za toho stavu, zvaná dle autora Armaniho zona. Nejčastěji však přichází v úvahu úplavice cukrová, u které se na změnách podílí i steatosa epitelií proximálních kanálků.
Výrazná akumulace glykogenu je také často spojená s některými nádory, a to tak, že přešla do jejich názvu klarocelulární nádory. Název je odvozen ze změněné tinkce následkem výrazné akumulace glykogenu (viz. výše). Jde např. o karcinom ledviny, některé kožní nádory jak z keratinocytů, tak z kožních adnex. V keratinocytech je často glykogen přítomen v nadměrném množství (zatím obr. 47)
Příčina nadměrné akumulace glykogenu za výše uvedených stavů není zcela jasná. Vše svědčí pro to, že nejde o projev primární nedostatečnosti glykogenolytických enzymů. Nedávné výzkumy ukazují velmi přesvědčivě, že regulátorem množství glykogenu mohou být i faktory jako buněčný objem a intracelulární pH. V experimentech s krysími hepatocyty bylo prokázáno, že zvětšení objemu hepatocytu, spojené se snížením osmotického tlaku, vede k výrazné aktivaci synthesy glykogenu při současném potlačení glykogenolysy. Podobný efekt vyvolalo snížení pH. Ukazuje se, že i nepříznivý metabolický stav (deplece ATP) může aktivovat mechanismy vedoucí k zvýšené synthese glykogenu (viz. aktivace protein dinasy zvýšenou koncentrací AMP). Výsledkem takových indukovaných stavů je tedy zmnožení granul glykogenu, jako výsledek změněné dynamiky jeho obratu v němž převládne synthesa (zdroj glukosy z glukoneogenese? a/nebo z extracelulárního prostoru pomocí transportérů glukosy).
Tento stav je nejčastěji způsoben fosforolytickou degradací glykogenu, vyvolanou potřebou glukosy jako energetického substrátu (hladovění, ischémie, porucha β oxidace). Redukce glykogenu může být dále způsobeno faktory, které jsou protějškem faktorů, vedoucích ke kompensačnímu zmnožení glykogenu, tj. hyperosmotickým zmenšením volumu buňky, zvýšení intracelulárního pH (viz. též výše). Totální trvalá absence glykogenu je způsobena deficiencí glykogen syntasy (viz. výše). Značný úbytek glykogenu může nastat při aktivaci autofagie.
4. Stavy spojené s deposicí abnormálního glykogenu (minimálně větveného,blízkého amylopektinu) ve formě tělísek, ustáleně zvaných polyglukanová nebo historicky corpora amylacea
Corpora amylacea (CA). Za některých stavů se glykogen akumuluje jako minimálně větvený polymer nejčastěji ve formě kulovitých tělísek, přirovnávaných k zrnku škrobu - corpora amylacea. Tato kulovitá tělíska (nebo méně pravidelná deposita), běžně nazývaná polyglukosanová (polyglucosan bodies), nebo též amylopektinová, jsou složena z abnormálně málo větveného glykogenu situovaného v základní cytoplasmě. CA mají velikost okolo 20 µm. Jsou amorfní v optickém mikroskopu. Mohou mít (v určitém stupni vývoje) dvojlom typu maltézského kříže a to i v parafinových řezech. V elektronovém mikroskopu mají dobře patrný vláknitý charakter, ale ultrastruktura nebývá zcela homogenní. Jsou přítomny i okrsky pleiomorfní naznačující účast degenerativních změn cytoplasmy (obr. 48). Obecně jsou považovaná za agresomy, v nichž je vedle abnormálního glykogenu segregována i řada proteinů, které jsou ubikvitovány a určeny k degradaci v proteasomu standardní cestou, která je však narušena a neefektivní. V mnoha případech byla pozorována aktivace lysosomálního systému.
Molekulární patogenesa CA však není velmi pravděpodobně jednotná. Je známo, že s jejich masivní deposicí těchto jsou spojeny zřejmě zcela metabolicky odlišné dědičné metabolické poruchy:
Laforova myoklonická epilepsie (autosomálně recesivní porucha) se projevuje generalizovanou deposicí CA s maximem v neuronech (obr. 49) a v dendritech, tedy zcela rozdílně od variant GSD IV, kde dominuje (mimo infantilní formy) axonální lokalisace . Byly prokázány dvě geneticky odlišné, ale fenotypově podobné (klinicky i na úrovni buňky) formy. U jedné ukázala vazebná analýza deficit laforinu, molekuly považované za fosfatasu, defosforylující glykogen (lokalisace v plasmatické membráně a v endoplasmatickém retikulu), u druhé mutaci v genu kodujicim E3 ubikvitin ligasu malin. Oba proteiny spolu za normálních okolností kooperují a jejich kooperací je údajně za inhibována synthesa glykogenu v neuronech (viz. výše). Laforovy epilepsie jsou neurologická onemocnění. Asymptomatická deposita tělísek jsou běžně přítomna např. i v hepatocytech a v kůži (v potních žlázkách, zejména apokrinních) a v dalších buněčných typech a to i ve větším množství. Je pozoruhodné, že se deposita neobjevují v astrocytech v množství vyšším, než odpovídá věku.
Zajímavé je pozorování Laforovy epilepsie, začínající jako progresivní hepatopatie s typickými tělísky jako prvý klinický projev nemoci.
Tělíska podobná Laforovým nebo přímo identická s nimi byla popsána v hepatocytech za různých stavů (viz. níže), aniž by šlo o systémové onemocnění.
Glykogenosa IV typu (deficit větvícího enzymu), maximálně se manifestující v játrech, i když tendence ke generalisaci je známa (viz. výše). Změny v játrech jsou demonstrovány na následujících obrázcích (obr. 50) (obr. 51)
Glykogenosa VII (deficit fosfofruktokinasy). U této glykogenosy byla popsána deposita polysacharidu polyglukanového typu (viz. výše)
Mutace AMPK (protein kinasa aktivovaná AMP) vedoucí k neregulované aktivitě této kinasy, což má za následek aktivaci celé řady procesů, včetně zvýšené resorpce glukosy aktivovaným GLUT-4 transportérem. To má za následek zvýšení synthesy glykogenu, pravděpodobně bez příslušné aktivace větvícího enzymu.
Dospělá forma polyglukosan body disease (APGBD) s různě intensivní a různě generalizovanou deposicí amylopektinových tělísek v periferních nervech, v CNS (axonech , astrocytech,male i volně v neuropilu) ve svalech, nerozlišitelná od adultní varianty GSD IV s prokazatelným enzymovým deficitem (viz. výše). Tyto změny provází neurologické poruchy (demence, ataxie) a periferní neuropatie, ale i viscerální poruchy na příklad kardiomyopatie.
Experimentálně vyvolaná zvýšená aktivita glykogen syntasy vedla ke generalizované deposici amylopektinových tělísek (viz. obr. 48). Vysvětlení je, že jde o relativní deficit větvícího enzymu, který nesleduje lineární deposici dostatečnou intensitou. Uvažuje se, že by bylo možno tímto způsobem vysvětlit některé stavy spojené s deposicí polyglukanových tělísek bez deficitu větvícího enzymu (viz. výše)
Po dlouhou dobu je známo, že se vedle zmíněných geneticky podmíněných metabolických poruch dochází k deposici CA pravidelně v centrálním nervovém systému ve výběžcích astrocytů, ale i ve výběžcích neuronů (CNS i sítnicových), ve kterých množství CA narůstá s věkem. Jejich největší koncentrace je zejména v subpiálních oblastech mozkové kůry (obr.52).Výskyt v astrocytech formálně odpovídá vysokému výskytu glykogenu v této glii, představující metabolickou reservu pro neurony. Nevylučuje se však ani původ mimo astrocyty. Tyto změny nevedou k neurologickým symptomům. K analogické deposici polyglukanových tělísek dochází v průběhu stárnutí s menší intensitou v kardiocytech ve vyšších věkových kategoriích, v axonech periferních nervů (obtížná dif. dg. od PGBD - viz. výše), ale i dalších orgánech
Přehled situací vedoucích k deposici CA je podána v následujícím obrázku 53.
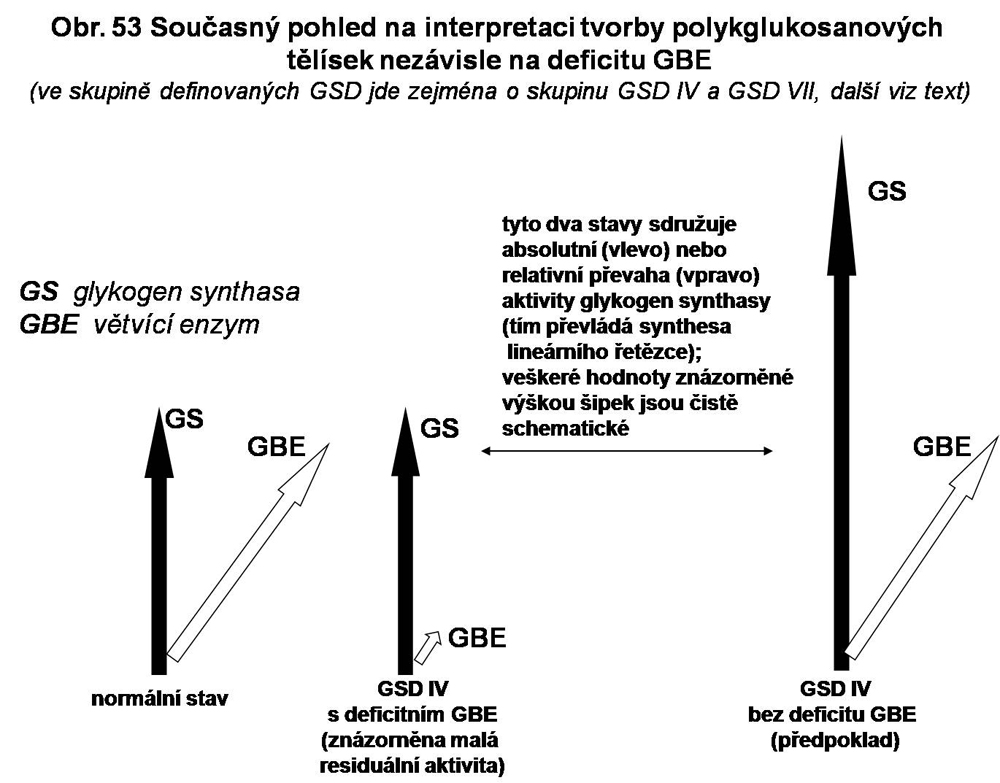
V současnosti jsou znalosti tohoto/těchto procesů minimální. Chybí i vysvětlení různě vyjádřeného toxického působení deponovaného minimální větveného glykogenu, která je velmi pravděpodobně velmi časným projevem poruchy a vede k segregaci a defektní clearance dalších cytosolárních proteinů. Pro identifikaci CA slouží i protilátka připravená proti polyglukanu Laforových tělísek, dostupná komerčně. CA jsou resistentní na natrávení diastasou (kompletně nebo zčásti). Údajně jsou totálně odstranitelné pektinasou.
Pektinasa používaná pro kompletní natrávení CA (viz. níže), je však definována jako galakturonidasa štěpící pektin - polymer galakturonové kyseliny.
CA vykazují velmi častou výraznou ubikvitinaci a deposici p62 proteinu. CA mají tak povahu agresomu. Je známo z několika studií, že deposice CA vede k částečné aktivaci lysosomálního systému. Velmi pravděpodobně jde o autofagií.
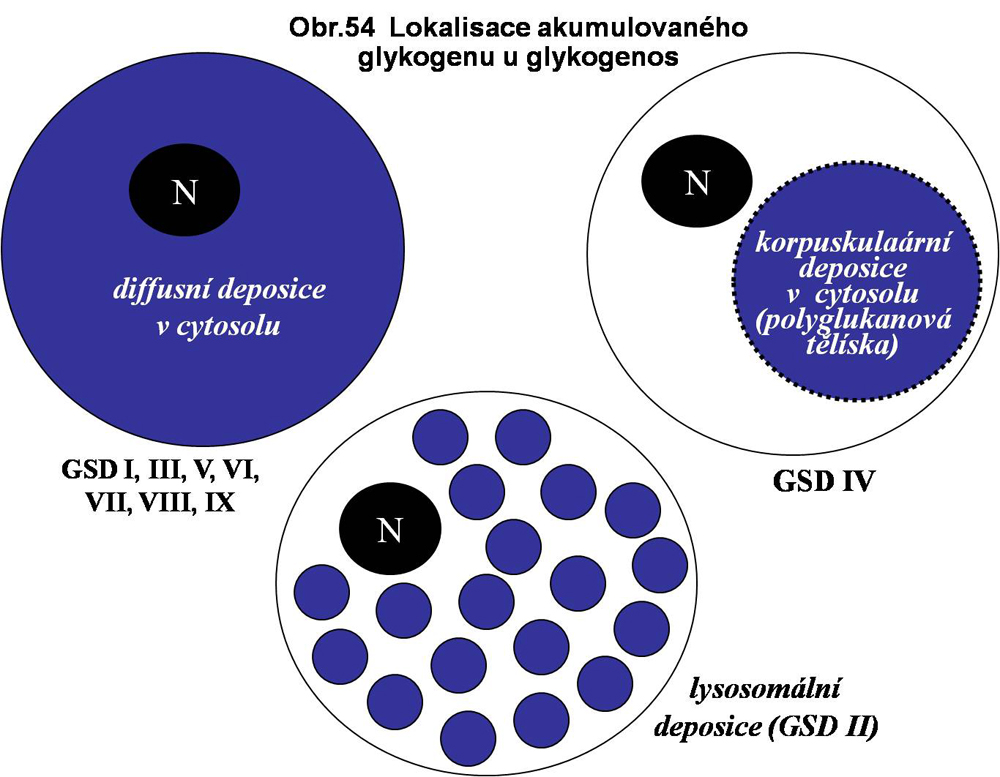
Subcelulární lokalisace akumulovaného glykogenu. Deposice glykogenu je u všech forem glykogenos lokalisována v cytosolu. Nejčastěji jde o difusní typ akumulace, v případě GSD IV je akumulace fokální/korpuskulární). Jedinou výjimkou je primárně lysosomálně lokalisovaná GSD II (obr. 54)
Pohled orgánový - obrat glykogenu v kritických orgánech postižených v rámci glykogenos; základní diferenciálně diagnostický pohled
Myokard. Kardiomyocyty obsahují standardně glykogen (agregáty typu β partikulí). Maximum glykogenu obsahují kardiocyty ve vodivých vláknech. Oba systémy vláken obsahují nejvíce glykogenu v časném postnatálním období. Běžný histologický obraz srdce v časném období po porodu se blíží glykogenose (světlá cytoplasma - viz. výše cytologický obraz buňky při nadměrné akumulaci glykogenu).
Celý systém synthesy glykogenu je definován: glykogenin a glykogen synthasa, představují isoformy více méně specifické pro svalový systém obecně; větvící enzym je sdílen se všemi ostatními tkáněmi.
Systém degradace glykogenu je znám jen částečně: fosforylasa je definována velmi povrchně - z literatury je známá jako "třetí typ" (vedle jaterní fosforylasy a fosforylasy kosterního svalu), podobně kinása fosforylasy.. Odvětvující enzym je sdílen se všemi ostatními tkáněmi
Srdce se nepodílí na regulaci hladiny krevního cukru (G6P je zpracován kardiomyocyty samotnými).
Postižení srdce v rámci generalizovaných glykogenos je známo u GSD II, III, IV.
U GSD IX (= deficity kinás fosforylasy ) nebylo postižení zatím popsáno. Výjimkou je infantilní varianta, která na rozdíl od ostatních glykogenos, postihuje masivně pouze srdce. Příčina, tedy zodpovědný enzym (fosforylasa, její specifická kinasa ) není však známa. Za zmínku stojí dysfunkce AMP aktivované minusy (mutace v gama 2 podjednotce) prokázaná u některých těchto vzácných infantilních kardiálních forem (důsledky postižení z dospělých viz. níže).
V diferenciální diagnose glykogenos je nutno počítat s více méně isolovaným postižení srdce (kardiomyopatie) bez výraznější známky generalizace metabolické poruchy u GSD III, GSD IV, u dospělých je to i dysfunkce AMPK (AMP aktivované protein kinasy) Situace je shrnuta na následujícím obrázkovém schématu (obr.55)

Biopsie srdce nepřichází v naprosté většině případě v úvahu. Vše je standardně řešeno biochemicky a DNA analysou. Pokud by k biopsii došlo, jsou
Ve všech případech je různě vyjádřená hypertrofie kardiomyocytů. Odlišení normálního množství glykogenu v kardiomyocytu od glykogenosy je poměrně snadné. Za normálního stavu v dospělosti je glykogen v malém množství přítomen zejména v elementech převodního systému, v kontraktilních kardiocytech je v malém množství mezi fibrilami a perinukleárně. V časných stadiích po porodu může být glykogen do značné míry přítomen i v kontraktilních kardiocytech a to ve značném množství.
Kosterní sval. Základní etapy procesu synthesy a degradace glykogenu jsou poměrně dobře definovány (obr.56).
Synthesa glykogenu: glykogenin type 1, glykogen synthasa typ 1 (svalové isoformy); větvící enzym je sdílen s ostatními buňkami; agregace je do β partikulí, lokalizovaných inter- a intramyofibrilárně, méně subsarkolemálně
Degradace glykogenu: fosforylasa (svalová isoforma), kinasa má svalově specifické isoformy podjednotek α a γ. Obecně sdílené jsou podjednotka β a kalmodulin. Je všeobecně přijímáno, že existují stále deficity kinás fosforylasy, které nejsou molekulárně geneticky definované, což svědčí o mechanismech aktivace fosforylasy, které čekají na objasnění. Značnou roli v degradaci může hrát proces autofagie.
Sval se nezúčastní regulace hladiny krevního cukru. G6P je tedy zpracován ve svalovém vláknu.

Postižení kosterního svalu (myopatie) jako výraz glykogenosy, nezávisle na věku je u GSD II, III, IV, V, VII, IX ( některé podtypy) a poruch v glykolytickém řetězci (obr.41)
Dospělé formy myopatií jako výraz GSD přichází v úvahu zejména GSD II ,GSD V, GSD VII (bývá spojena s tendencí k hemolyse - viz. text výše) , GSD IX (deficity podjednotek kinás fosforylásy)
Nálezy ve svalové biopsii
Za normálních okolností je glykogen se vláknech kosterního svalu lokalizován v mírně variabilním množství v sarkoplasmě mezi fibrilami (inter- a intramyofibrilárně), mírně subsarkolemálně.
Pro diagnosu je rozhodující je biochemické vyšetření vzorku svalu. Deficit kinasy fosforylasy je prokazatelná údajně i v erytrocytech
Obě cesty obratu glykogenu jsou dnes poměrně dobře definovány (obr.57).
Synthesa glykogenu: glykogenin typ 2, glykogen sythasa typ 2 (orgánově omezené isoformy), větvící enzym, sdílený s ostatními buňkami organismu.
Degradace glykogenu: fosforylasa (orgánově specifická isoforma), kinasy fosforylasy (orgánově specifické podjednotky α a γ, společná podjednotky β a kalmodulin). Významnou složkou degradace může být autofagocytosa.
Hepatocyty mají zásadní význam v regulaci hladiny krevního cukru. Mají aktivitu G6Pasy, uvolňující štěpící G6P uvolněný z glukoneogenese nebo glykogenolýsy. Uvolněná glukosa je pomocí GLUT 2 transporteru předávána na krevním pólu hepatocytu do oběhu. To je predisponuje k postižení u GSD I a GSD XI

Postižení hepatocytů se projevuje prakticky u všech typů glykogenos nezávisle na věku. Jde zejména o GSD I, II, III, IV, VI, některé formy GSD VIII, IX (nejasná klasifikace, viz. text), dále GSD X, XI a poruchy glykolytického cyklu (obr.41)
Výjimkou jsou enzymy, jejichž isoformy jsou čistě svalové: GSD V, některé GSD IX
Postižení v dětském věku se známkami selhání orgánu je známé u GSD IV, a u některých variant GSD III (GSDIIIb)
Játra jsou postižena i v rámci Laforovy epilepsie (i když prakticky vždy asymptomaticky, blíže viz. výše u Laforovy epilepsie). Biopsie jater patří k diagnostickým přístupům. V hepatocytech lze očekávat řadu sekundárních poruch obratu glykogenu ať již u genetických poruch jiných metabolických cest, tak v rámci poruch získaných (viz. výše)
V ledvinách jsou s glukosou - základním stavebním kamenem glykogenu spojeny tři odlišné procesy:
(i) intensivní transcelulární transport z apikálního směrem k pólu basolaterálním a odtud do intersticia. Tento transport je lokalizován do proximálních kanálku, bohatě vybaveného kartáčovým lemem na apikálním pólu Zde je lokalisován "sodium glucose transporter" (SGLT 1 a 2), spřažující transport glukosy s transportem Na+ (Na+-K+-ATPAsa).Basální část s výrazným membránovým labyrintem zprostředkuje přenos glukosy usnadněnou difusí pomocí Glu-2 transportéru (dysfunkční u Fanconi-Bickelova syndromu).V prvé části proximálního tubulu (pars konvoluta) je zejména SGLT2 transportér (vysoko kapacitní, ale nízkoafinitní pro glukosu; přenos glukosy a |NA+ je v poměru 1:1). V následných částech prox. tubulu, zejména v pars recta) je SGLT1 transportér (vysoko afinitní, ale málo kapacitní, přenášející glukosu a Na+ v poměru 2:1), který resorbuje zbytek glukosy z glomerulárního filtrátu. Cca 98% nálože v glomerulárním filtrátu je resorbováno v proximálním tubulu. Zbytek, cca 2% je reabsorbována v pars recta proximálního tubulu kde je SGLT1. Za fyziologických podmínek se do Henleovy kličky už žádná glukosa nedostává. Za normální hladiny glykémie je veškerá glukosa resorbována.
(ii) syntesa glykogenu vyjádřená prakticky ve všech oblastech kanálkového epitelu. Zde představuje glykogen klasickou energetickou reservu
(iii) pouze v epiteliích proximálního kanálku slouží glykogen i jako reservoár glukosy k spoluregulaci hladiny krevního cukru. Tyto oblasti se vyznačují expresí G6Pasy, sloužící pro uvolnění glukosy, před jejím transportem do krve na basálním pólu epitelu.
Řada studií svědčí však pro to, že v normálních ledvinách jsou minimální množství glykogenu, na rozdíl od jater, kosterního svalu a srdce a dalších orgánů.
Synthesa glykogenu v ledvinách je realisována glykogeninem a glykogen synthasou, oba typy 1 (tzv. svalové) a větvícím enzymem, který je společný všem tkáním
Degradaci glykogenu zprostředkuje fosforylasa patřící do tzv. třetího typu ("mozková" isoforma, odlišná od jaterní a svalové, viz. shora). Kinasu fosforylasy lze pravděpodobně řadit do "svalové" skupiny. Lysosomální degradace je vyjádřená.
Hyperglykemické stavy, zvyšující nálež glukosy v glomerulární filtrátu, která přesahuje transportní kapacitu proximálního tubulu. Glykogen je za těchto stavů akumulován v pars recta - Armaniho, nebo též Ebstein-Armaniho zona). Stejně tak primární defekty resorpce v proximálním kanálku. (Fanconiho syndrom) . U diabetu se poslední dobou se zdůrazňuje i vysoký podíl steatosy v pars recta.
Poznámka. Obtížně lze hledat vysvětlení pro akumulaci glykogenu na podkladě zvýšené nálože glukosy, jejíž resorpce má překročenou kapacitu SGLT transportérů (viz. výše).
Historie Armaniho-Ebsteinovy zony. Armani se domníval, že k akumulaci glykogenu dochází v sběracích kanálcích, následně podle Ebsteina šlo o Henleho kličku. Teprve pozdější studie ukázaly, že jde o pars recta proximálního tubulu. Původní nálezy tak byly korigovány.
Na rozdíl od hyperglykemií u člověka se v experimentálním diabetu u krys glykogen akumuloval v části distálních tubulů, později i v proximálních kanálcích. V jiných podobných pokusech se glykogen akumuloval dominantně v ascendentní Henleho kličce. V poslední době se stále více mluví o poškození Henleho kličky u lidského diabetu, projevující se poklesem močového uromodulinu, který v této části nefronu produkován.
U Fanconiho-Bickelova syndromu (GSD XI, viz. výše), podmíněného defektem Glu-2 přenašeče na basálním pólu buněk proximálního kanálku, dochází k akumulaci glykogenu též v proximálním kanálku, převážně v p.recta.
U glykogenosy typ I je glykogen akumulován v celém rozsahu proximálního kanálku, tedy logicky v místech exprese G6Pasy.
U GSD II dochází k lysosomálnímu střádání v glomerulu a v nefronu mimo proximální kanálky
Studie postižení ledvin u GSD IV jsou zcela ojedinělé. Pokud byly ledviny vyšetřeny, byla deposita popsána v Henleho kličce a ve sběracích kanálcích.
O akumulaci glykogenu u ostatních forem glykogenos nejsou údaje.
Neurony. Biologie glykogenu v neuronech se jeví podle posledních studií jako zcela odlišná od ostatních buněk v těle.
Pokud jde o synthesu glykogenu (GS svalového typu), je v neuronech trvale blokována (viz. citaci) permanentní cílenou degradací glykogen syntásy a proteinu, který podmiňuje její aktivaci (PTG protein targeting to glycogen - protein, který cílí protein fosfatasu 1 ke glykogen synthase, sloužící k její aktivaci). Tato trvalá degradace (v proteasomu) je zprostředkována komplexem malin a laforin (viz. výše Laforovu epilepsii). Z dosavadních studií vyplývá, že aktivita větvícího enzymu je nízká (nejsou kvantitativní údaje).
Aktivita fosforylasy není údajně detekovatelná. O odvětvujícím enzymu nejsou údaje k disposici. Centrem metabolismu glykogenu jsou astrocyty, které jsou v tomto smyslu velmi aktivní a kompletně vybavené. Obrat glykogenu v nich je do znané míry regulován neurony. Produkty degradace glykogenu a glykolýzy v astrocytech slouží jako energetický substrát pro neurony.
Intensivní obrat glykogenu je kompatibilní s častým výskytem corpora amylacea (viz. výše)
ReferenceTyto nálezy mohou vysvětlit relativní resistenci neuronů k postižení ve skupině glykogenos. Existují následující výjimky.
I přes názor, že synthesa glykogenu představuje ohrožení integrity neuronu (viz. úvod a níže) a že je tedy inhibovaná, lze z přítomnosti neuronálního střádání u infantilní formy GSD II (Pompeho nemoc, viz. výše) usoudit, že k určitému glykogenu obratu dochází.
Situace u Laforovy epilepsie je specifická a stojí zcela mimo klasické GSD.
Postižení v rámci glykogenos sekundárního typu při poruchách glykolýsy (viz. obr. 41) se může manifestovat pouze následky glykolýzy samotné.
Pokud jde o infantilní form GSD IV (deficit větvícího enzymu) k manifestaci nemoci buď v neuronálních perikaryiích nedochází a projevy (deposice corpora amylacea) pouze ve výběžcích a situace tak formálně připomíná Alexandrovu nemoc (mutace GFAP). Některá publikovaná pozorování však popisují deposici corpora amylacea i v neuronech zejména míchy a kmene mozkového.
GSD II a GSD IV jsou tak jediné glykogenosy (nepočítáme-li glykogenosy z poruchy glykolýzy) které mohou postihovat CNS a vést k neurologické poruše.
Při hodnocení postižení CNS je nutno si uvědomit, že vedle relativně inertních neuronů (výjimkou je Laforovy epilepsie) je masivní postižení astrocytů (centrum obratu glykogenu v CNS) možné a může zodpovídat za neurologickou poruchu.
Souhrn orgánové patologie glykogenosy je podán na obr.58
