Bipolární forma - střídání deprese a mánie.
Unipolární forma - pouze jedna fáze (obvykle depresivní).
Těžká podoba postihuje asi 1% populace a je obvykle dobře léčitelná. Příčiny bipolárního onemocnění jsou zřejmě do značné míry genetické, i když odpovídající geny nebyly dosud nalezeny. Anatomické změny v mozku jsou podobné jako u chronické schizofrenie. Protože existují i smíšené psychózy, tj. s příznaky schizofrenie i maniodepresivity, vznikla domněnka, že schizofrenie a maniodepresivní psychóza mohou být dva projevy téhož postižení funkčních nervových soustav.
Na léčbu antidepresivy odpovídá 65-70% pacientů. Zlepšení nastává po asi 10 dnech farmakoterapie a je úplné po asi 8 týdnech. Elektrokonvulzivní terapie je účinná v dalších 10-15%. Tedy asi 20% depresivních pacientů je rezistentních ke všem známým formám terapie. Neléčená deprese vede ve 25-30% k pokusu o sebevraždu.
Biologická psychiatrie se snaží popsat a vysvětlit vznik a léčbu afektivních poruch na základě poznatků získaných jak klinickým hodnocením, tak měřením biochemických a fyziologických změn. Orientace na výsledky získané metodami různých přírodních věd neznamená popření psychoterapeutických přístupů k těmto duševním poruchám, nýbrž pouze a jen zaměření na parametry měřitelné a ověřitelné současnými biologickými, chemickými a fyzikálními metodami. Tomu odpovídají i příslušné hypotézy o biologické podstatě afektivních poruch.
Z biochemického a elekrofyziologického hlediska existuje řada důkazů pro to, že na vzniku afektivních poruch se podílejí změny v přenosu nervového signálu, především v oblasti synapsí. Svědčí pro to jednak změny koncentrací neuromediátorů a jejich metabolitů v mozku, mozkomíšním moku a plazmě u některých depresivních pacientů, jednak dosud známé účinky antidepresiv. Ze zřejmých etických důvodů je studium přenosu signálu a receptorových a postreceptorových procesů při afektivních poruchách a jejich terapii zatím omezeno na použití nepřímých postupů, tj. na měření změn v lidských mozcích post mortem nebo experimenty s použitím zvířecích a buněčných modelů (leukocyty, trombocyty, buněčné kultury, liposomy, atd.).
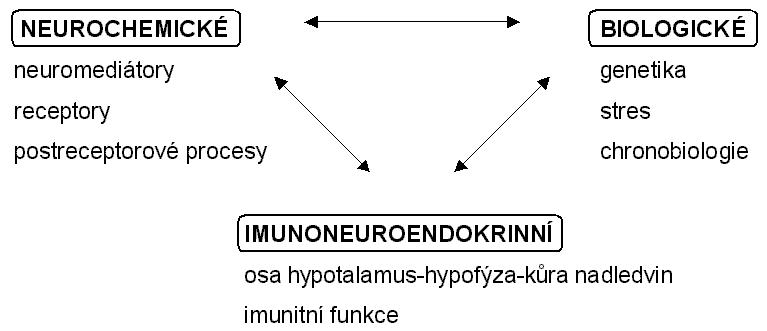
Zdroje poznatků vedoucích k formulaci hypotéz afektivních poruch se nacházejí především v oblasti biologické (genetické faktory, účinky stresu, chronobiologické aspekty), imunoneuroendokrinní (změny aktivity osy hypotalamus-adenohypofýza-kůra nadledvin, uvolňování cytokinů apod.) a neurochemické (narušení synaptického přenosu signálu). Toto rozdělení vychází především z klasického rozdělení biomedicínských oborů a metod výzkumu používaných pro studium systémů ovlivněných při afektivních poruchách a jejich terapii. Je zřejmé, že uvedené oblasti jsou navzájem propojeny. Důkazy pro existenci biochemických základů afektivních poruch byly proto od počátku hledány především:
ve funkci neuromediátorových systémů;
v genetických faktorech;
v neuroendokrinních změnách.
PŘÍSTUPY KE STUDIU AFEKTIVNÍCH PORUCH
Biologické modely afektivních poruch vycházejí především z vlivu genetických nebo stresových faktorů na vznik a průběh onemocnění. Tyto faktory mohou přes imunoneuroendokrinní a neurochemické účinky ovlivňovat přenos signálu v CNS a vést ke vzniku afektivních poruch.
Bylo potvrzeno, že existuje vrozená náchylnost pro vznik afektivních poruch, ale dosud se nepodařilo spolehlivě identifikovat příslušný gen. Genetické studie uvádějí hodnoty relativního rizika (poměr rizik pro příbuzné nemocných a pro kontrolní osoby) od 10 do 15 pro bipolární poruchu a od 2 do 5 pro depresi. Shoda výskytu maniodepresívního onemocnění je u monozygotních dvojčat kolem 57 % u dizygotních 14 %. Geneticky podmíněnou zvýšenou náchylnost ke vzniku afektivních poruch potvrzují také adopční studie. Nicméně je zřejmé, že genetické faktory samy o sobě nejsou postačující pro vznik onemocnění a že jejich úloha a přenos nejsou dostatečně známy. Určité problémy způsobuje genealogickým studiím skutečnost, že diagnóza onemocnění vychází pouze z klinických údajů (neexistuje vhodný biologický test nebo znak), přičemž diagnostická kritéria se vyvíjejí a mění.
První genetické modely bipolární poruchy předpokládaly dominantní pohlavně vázaný přenos (chromosom X), což se však ukázalo jako nepravděpodobné. Na základě zřejmé heterogenity bipolárního onemocnění se uvažuje spíše o přenosu podmíněném více geny (oligogenní přenos). Tyto geny však nebyly dosud nalezeny. Předpokládá se, problémy v určení podílu genetických faktorů na vzniku bipolání poruchy jsou způsobeny heterogenitou onemocnění. Je možné, že pacienti s časným začátkem onemocnění nebo odpovídající na léčbu lithiem tvoří podskupinu vyšším genetickým rizikem. Vznikly hypotézy, podle nichž existuje plynulý, geneticky podmíněný přechod od unipolárního depresivního onemocnění, přes bipolární afektivní poruchu a schizoafektivní onemocnění, až k různým formám schizofrenie. Jiné studie však podporují hypotézu nezávislosti afektivních poruch a schizofrenie, nebo předpokládají jen částečný překryv genotypů bipolárního onemocnění a schizofrenie.
Genetické faktory mohou ovlivňovat náchylnost ke vzniku afektivní poruchy tím, že se podílejí:
na zvýšení citlivosti jednotlivých osob k opakujícím se událostem indukujícím depresi;
na nedostatečné funkci určitých homeostatických mechanismů.
Je zřejmé, že geneticky podmíněná náchylnost
ke vzniku afektivní poruchy může být výrazně ovlivněna různými vývojovými
faktory a vlivem okolního prostředí. Podstatná může být indukce či
suprese přepisu genetické informace genů kódujících proteiny, které se
podílejí na plasticitě synapsí a růstu neuronů.
Stres má vliv
na průběh afektivní poruchy a předpokládá se, že se podílí i na jejím
vzniku. Mechanismy účinků stresu propojují přístupy biologické,
imunoneuroendokrinní a neurochemické. Při stresu je např. indukována
exprese stresových proteinů a je zvýšena biosyntéza polyaminů. Vznikla
hypotéza, podle níž polyaminy a změny v jejich metabolismu mohou být
zahrnuty v patofyziologii afektivních poruch. Opakovaným stresem mohou být
vyvolány změny ve funkci transkripčních faktorů, což může vést k neurobiologickým
změnám v limbickém systému a zvyšovat tak náchylnost pacientů k dalším
depresivním epizodám. Po chronickém stresu bylo pozorováno např. zkrácení
dendritů a úbytek jejich průměrného počtu u pyramidových neuronů v hipokampu.
Neurochemický přístup k afektivním poruchám vychází z předpokladu, že příčinou jejich vzniku je narušení přenosu a zpracování signálu v CNS. Předpokládáme-li, že myšlenky a pocity souvisejí se zvýšenou či sníženou aktivací určitých skupin neuronů, potom jejich ovlivnění je možné především v oblasti synapsí, které zajišťují kovergenci a divergenci nervových signálů. Na základě mechanismů účinků psychoaktivních látek lze usoudit, že k poruchám může dojít především při transdukci signálu, tj. změně šířícího se akčního potenciálu na procesy vyúsťující v uvolňování neuromediátorů do štěrbiny s následnou aktivací pre- nebo postsynaptických receptorů a příslušných postreceptorových změn. U receptorových systémů, které se pravděpodobně podílejí na vzniku afektivních poruch a na terapeutických účincích antidepresiv, se jako transdukční prvek (protein zprostředkující transmembránový přenos a zesílení signálu o navázání neuromediátoru k receptorovému vazebnému místu) uplatňuje obvykle určitý G protein. Tento transdukční prvek převádí signál na efektorový systém, kterým je obvykle buď iontový kanál řízený G proteinem, nebo enzymový systém katalyzující vznik druhých poslů. Proteinkinasy aktivované druhými posly potom katalyzují fosforylaci iontových kanálů, vazebných míst, G proteinů, adenylátcyklas a jiných enzymů, což vede ke změnám vlastností buňky.
Trandukce nervového signálu v synapsi tedy zahrnuje řadu poměrně složitých procesů, které jsou za normálních podmínek v dynamické rovnováze. Neurochemické hypotézy afektivních poruch předpokládají, že narušení této rovnováhy vede k dlouhodobému ovlivnění neuronální aktivity v určitých oblastech mozku odpovědných za vznik pocitů. Prvotním předpokladem je, že deprese souvisí se sníženou a mánie se zvýšenou aktivitou těchto mozkových oblastí.
Schopnost neuronů reagovat na přicházející akční potenciály nespočívá pouze ve vzniku excitačních nebo inhibičních postsynaptických potenciálů, ale rovněž v modifikaci citlivosti, s níž neurony na vnější podněty reagují (přes fosforylaci a defosforylaci membránových i nitrobuněčných proteinů, přes ovlivnění genové exprese molekul podílejících na transmembránovém přenosu signálu, atd.). Výsledná odezva neuronů na přicházející impulsy je velmi komplexní, neboť zahrnuje řadu křížových propojení a záporných zpětných vazeb. Nelze tedy jednoduše říci, že zvýšené excitační nebo inhibiční vstupy mají jednoznačný výsledek na vlastnosti daného neuronu. Dochází tak jevům, kdy např. po dlouhodobě zvýšené aktivaci určitých buněčných procesů dojde, při nezměněných vstupech, k jejich poměrně rychlé deaktivaci. Není zatím známo, zda takové změny na molekulární úrovni přímo souvisejí se změnami nálady.
Neurochemické
faktory sledované při afektivních poruchách
|
neuromediátory |
dostupnost |
|
|
rychlost metabolismu |
|
receptory |
počet nebo hustota |
|
|
afinita |
|
|
senzitivita |
|
postreceptorové systémy |
počet a aktivita G proteinů |
|
|
efektorové enzymy |
|
|
systémy 2. poslů |
|
|
proteinkinázy |
|
|
fosfatázy |
|
|
transkripční faktory |
Při afektivních poruchách jsou pozorovány změny aktivity osy hypotalamus-hypofýza-kůra nadledvin (HPA) nebo také osy hypotalamus-hypofýza-štítná žláza. Hlavní pozornost je věnována uvolňovacímu faktoru pro kortikotropin (CRF) a tedy procesům v ose HPA:
neurony v hypotalamu uvolňují CRF;
CRF se dostává krví do adenohypofýzy a stimuluje uvolňování adrenokortikotropního hormonu (ACTH);
ACTH stimuluje uvolňování glukokortikoidů (především kortizolu a kortikosteronu) a jiných steroidů z kůry nadledvin;
ACTH zpětnovatebně inhibuje vylučování CRF;
kortikosteroidy zpětnovazebně inhibují uvolňování CRF z hypotalamu (přes odpovídající receptory) i ACTH z adenohypofýzy.
U více než 50% depresivních pacientů je mírně zvýšena funkce osy HPA indikovaná např. zvýšenými koncentracemi CRF v mozkomíšním moku (CSF). Vyšší aktivita osy HPA v těžké depresivní fázi se projevuje také méně výrazným snížením plasmatických koncentrací ACTH a kortisolu po podání dexametasonu. Dexametasonový supresní test (DST) byl navržen jako specifický test pro diagnózu těžké depresivní fáze, specifičtější je však kombinovaný dexametasonový-CRF test, kdy je nejprve podán dexametason a po určité době CRF. Zvýšená odezva osy HPA v tomto testu byla pozorována u více než 80% pacientů v těžké depresivní fázi.
Další pozorování
ukázala, že počet vazebných míst pro CRF ve frontální kůře sebevrahů
je snížen, pravděpodobně v důsledku dlouhodobě zvýšené dostupnosti
CRF, že podání CRF zdravým dobrovolníkům vyvolává příznaky depresivní
poruchy a že je snížená odezva (měřená uvolňováním ACTH) na podání
CRF depresivním pacientům způsobená pravděpodobně zvýšenými základními
hladinami kortisolu.
Neuroendokrinní
hypotéza afektivních poruch: endokrinní
abnormality spojené s depresí jsou výsledkem zvýšeného uvolňování CRF v
hypotalamu i mimo něj.
Hypotéza o narušené glukokortikoidní zpětné vazbě při depresi: nedostatečné zpětnovazebné působení kortizolu při depresi je způsobeno sníženou hustotou kortikosteroidních receptorů v hipokampu a v hypotalamu, tedy pravděpodobně poruchou v regulaci genové exprese těchto receptorů.
Poznatky z psychoneuroimunologie ukazují, že deprese a život ve stresu mohou měnit imunitní funkce. Těžká depresivní fáze může být doprovázena aktivací imunitního systému nebo zánětlivou odezvou se zahrnutím fagocytujících buněk (monocytů, neutrofilů), aktivace T buněk, proliferace B buněk, změnou hladin proteinů akutní fáze, vyššího titru protilátek (antijaderných, antifosfolipidových), zvýšené sekrece prostaglandinu, poruch u exopeptidasových enzymů a zvýšené produkce interleukinů (IL-1b, IL-6) mononukleárními buňkami v periferní krvi. Změny v těchto parametrech jsou ale menší, než při imunitních poruchách.
Imunoneuroendokrinní a neurochemické přístupy ke studiu afektivních poruch jsou propojeny přes vzájemné ovlivňování funkcí monoaminergních neuromediátorových systémů a osy HPA. Existují důkazy svědčící pro to, že CRF účinkuje jako neuromediátor v locus coeruleus (LC, hlavní zdroj noradenregních neuronů) při stresem indukované aktivaci LC. Bylo zjištěno, že jak katecholaminy, tak kortikosteroidy modulují funkci b-adrenoceptorů a to zřejmě v opačném smyslu. Byla zjištěna snížená neuroendokrinní odezva (měřená uvolňováním prolaktinu) po dodání tryptofanu depresivním pacientům. Další prekursor serotoninu, 5-hydroxytryptofan, zvyšuje hladiny ACTH a kortisolu více u depresivních pacientů, než u kontrol. Hypotermická odezva vyvolaná specifickou aktivací presynaptických 5-HT1A receptorů je při depresi snížená.
Cytokiny mají klíčovou úlohu v imunitní aktivaci a ovlivňují rovněž centrální nervový systém. Předpokládá se, že zvýšená aktivita osy HPA pozorovaná často při těžké depresi je důsledkem přímého stimulačního působení cytokinů na hypotalamus. Existuje také propojení mezi cytokiny a katecholaminy; např. NA stimuluje uvolňování IL-6 z astrocytů a vylučování dalších interleukinů vztažených k IL-6. Lze tedy předpokládat, že NA uvolňovaný při stresu aktivuje cytokiny a vede k imunitním jevům spojeným se stresem.
Iterleukinová hypotéza deprese: změny v buněčné a humorální imunitě a v sekreci prostaglandinu při depresi mohou být vysvětleny mechanismy vztaženými k nadměrnému uvolňování IL-1b a IL-6. Tyto interleukiny patří mezi hlavní imunitní a zánětlivé mediátory. Celková imunitní aktivace může způsobit snížení dostupnosti plasmatického L-tryptofanu (L-TRP), který je prekursorem serotoninu (5-HT) nebo může být zvýšená sekrece imunitních složek vztažena k hyperaktivitě osy HPA a zvýšená aktivita osy HPA může mít záporné zpětnovazebné účinky na imunitní systém.
|
leukocyty, T buňky |
|
zvýšený
počet leukocytů - neutrofilů a monocytů, tj. fagocytujících buněk |
|
zvýšený poměr CD4+/CD8+
(T helper/T suppressor) - určen částečně zvýšením počtu a
procenta CD4+ buněk |
|
aktivace T buněk - zvýšený
počet a procento CD25+ a HLA-DR+ buněk, vyšší
hladiny rozpustných IL-2 receptorů |
|
interleukiny |
|
vyšší produkce IL-1b
a IL-6 při mitogenní stimulaci mononukleárních buněk z periferní
krve |
|
glukokortikoidní rezistence
vztažená k vyšší produkci IL-1b a IL-6, nebo ke snížení
glukokortikoidních receptorů na lymfocytech při depresi |
|
B buňky, autoimunita |
|
proliferace
B buněk |
|
vyšší
titry antijaderných autoprotilátek |
|
zvýšená aktivita
antifosfolipidových (antikardiolipin, antifosfatidylserin) autoprotilátek |
|
vyšší
hladiny IgG v plasmě |
|
proteiny akutní fáze |
|
vyšší plasmatické
hladiny pozitivních proteinů akutní fáze - haptoglobinu,
ceruloplasminu, hemopexinu a a1-antitrypsinu |
|
nižší plasmatické
hladiny negativních proteinů akutní fáze - transferrinu, albuminu,
proteinu vážícího retinol |
|
peptidasy |
|
nižší
aktivita sérové dipeptilpeptidasy IV |
|
nižší
aktivita angiotenzin konvertujícího enzymu |
(podle Maes M.: Prog. Neuro-Psychopharmacol. & Biol. Psychiat. 19, 11-38, 1995)