Mood disorders are characterized by depression, mania, or both. In this chapter, biochemical hypothesis of affective disorders are presented. Classification, clinical description and criteria for diagnosis of disorders of mood are not described.
Depression and mania are thought to be heterogeneous illnesses that can result from dysfunction of several neurotransmitter or metabolic systems. Approaches of biological psychiatry to the affective disorders are summarized in Table 5.1.
Table 5.1 Biological psychiatry and affective disorders
|
BIOLOGY |
genetics |
vulnerability to mental disorders |
|
stress |
increased sensitivity |
|
|
chronobiology |
desynchronisation of biological rhythms |
|
|
NEUROCHEMISTRY |
neurotransmitters |
availability, metabolism |
|
receptors |
number, affinity, sensitivity |
|
|
postreceptor processes |
G proteins, 2nd messengers, phosphorylation, transcription |
|
|
IMMUNONEURO- ENDOCRINOLOGY |
hypothalamic-pituitary-adrenocortical system |
increased activity during depression |
|
immune function |
different changes during depression |
Theoretical and clinical studies have provided evidence that the monoamine neurotransmitter systems are involved in the treatment of affective disorders. These studies have led to a series of hypotheses concerning the mechanism of the action of antidepressant treatments, as well as pathophysiology of depression, that have focused on alterations in brain levels of serotonin and norepinephrine or their receptors.
Studies at the level of neurotransmitters and receptors have not generated a compelling model of antidepressant action or the pathophysiology of depression. For example, common action of antidepressant treatments at the level of monoamines or their receptors was not identified so far. It is possible that there is more than a single mechanism by which antidepressant treatments exert their therapeutic actions.
An updated hypothesis suggests that postreceptor intracellular targets mediate the long-term, therapeutic action of antidepressant treatments.
5.1. Neuroendocrine hypotheses
Changes in the activity of the hypothalamic-pituitary-adrenal (HPA) axis or hypothalamic-pituitary-thyroid (HPT) axis were observed during depression. An increased function of the HPA axis was described in more than 50% of depressed patients. Abnormalities of the HPA axis in patients with depression are summarized in Table 5.2.
The dexamethasone suppression test (DST) is used to determine the sensitivity of the HPA axis to negative feedback. 1 mg of dexamethasone is usually administered at 23:00 h and plasma cortisol is monitored following morning. In healthy subjects dexamethasone suppresses plasma cortisol. DST was suggested as test for diagnosis of major depressive disorder, since increased function of HPA axis can be expressed in reduced decrease of ACTH and cortisol levels after dexamethasone administration. But it was demonstrated that DST test is unsufficiently specific.
Table 5.2. Abnormalities of the hypothalamic-pituitary-adrenal (HPA) axis in patients with depression
|
cortisol hypersecretion |
|
increased urinary free cortisol |
|
increased CSF corticotrophin releasing factor |
|
increased circulating ACTH |
|
abnormal circadian rhythms of cortisol |
|
abnormal dexamethasone suppression |
|
decreased glucocorticoid receptor sensitivity |
|
decreased release of ACTH to CRF |
|
increased adrenal gland size |
CSF – cerebrospinal fluid, ACTH – adrenocorticotrophic hormone, CRF – corticotrophin-releasing factor
(See: Trimble M.R.: Biological Psychiatry. 2nd ed., John Wiley & Sons, New York, 1996.)
Neuroendocrine hypothesis of affective disorders were formulated on the base of abnormalities in HPA axis function and disturbed glucocorticoid feedback in depression (Table 5.3).
Table 5.3. Neuroendocrine hypotheses of affective disorders
|
Endocrine abnormalities associated with depression are results of increased releasing of CRF both in hypothalamus and out of them. |
|
Disturbed glucocorticoid feedback in depression is caused by lower density of corticosteroid receptors in hippocampus and hypothalamus during depression (i.e. by disturbed regulation of gene expression of these receptors). |
5.2. Neurochemical hypotheses
There are evidences that signal transmission through chemical synapse is disturbed in the affective disorders. Changes in neurotransmitters, membrane receptors and postreceptor pathways are studied during mental illness and its treatment. The neurochemical parameters studied in affective disorders are summarized in Table 5.4.
Table 5.4. Neurochemical parameters studied in affective disorders
|
neurotransmitters |
availability |
|
metabolism |
|
|
receptors |
number or density |
|
affinity |
|
|
sensitivity |
|
|
postreceptor systems |
number and activity of G proteins |
|
effector enzymes |
|
|
2nd messengers systems |
|
|
protein kinases |
|
|
phosphatases |
|
|
transcription factors |
5.3. Neurotransmitter hypotheses
Discovery of the tricyclic antidepressant drugs supported hypothesis about significant role for the biogenic amine, particularly NE and 5-HT in the ethiopathogenesis of affective disorders. Reduced activity of the serotonergic and noradrenergic systems has been reported in subgroups of patients with major depression, but this has not been observed in all depressed patients.
Initial neurotransmitter hypotheses were supported by effect of tricyclic antidepressants on reuptake of NE and 5-HT, effect of monoamine oxidase inhibitors (MAOI) on catabolism of monoamine neurotransmitters and effect of reserpine on vesicular transport of neurotransmitters (Table 5.5). Reuptake inhibition results in an enrichment of the NE and 5-HT in the synaptic cleft. Similar effect can be achieved with MAO inhibitors. For many years this effect was considered to be the precondition for antidepressant activity. The first major theory about the biological ethiology of depression was monoamine hypothesis supposing that:
Depression was due to a deficiency of monoamine neurotransmitters, norepinephrine and serotonin. MAOI act as antidepressants by blocking of enzyme MAO, thus allowing presynaptic accumulation of monoamine neurotransmitters. Tricyclic antidepressants act as antidepressants by blocking membrane transporters ensuring reuptake of 5-HT or NE, thus causing increased extracellular neurotransmitter concentrations.
Table 5.5. Data for neurotransmitter hypothesis
|
|
|
In addition to the role of noradrenergic and serotonergic systems in depression, cholinergic and dopaminergic and others systems were considered, so, many neurotransmitter hypotheses were formulated (Table 5.6).
Table 5.6. Neurotransmitter hypothesis of affective disorders
|
catecholamine hypothesis |
|
indolamine hypothesis |
|
cholinergic-adrenergic balance hypothesis |
|
„permissive“ hypothesis |
|
dopamine hypothesis |
|
hypothesis of biogenic amine |
|
monoamine hypothesis |
Permissive biogenic amine hypothesis persists as part of more recent hypothesis:
A deficit in central indolaminergic transmission permits affective disorder, but is insufficient for its cause; changes in central catecholaminergic transmission, when they occur in the context of a deficit in indoleaminergic transmission, act as a proximate cause for affective disorders and determine their quality, catecholaminergic transmission being elevated in mania and diminished in depression.
However, neither the catecholamine nor the serotonin hypothesis could be confirmed in depressive patients. Clinically, the onset of the antidepressant action differs from the biochemical effects; the uptake inhibition occurs suddenly, whereas the therapeutic effect needs two or three weeks of pharmacotherapy. Furthermore, depletion of 5-HT or NE in healthy individuals does not induce clinically significant depressive symptomatology. These are reasons why receptor and postreceptor events are observed during affective disorders and their treatment at present time.
5.4. Receptor hypotheses
The neurotransmitter receptor hypothesis posits that disturbance occurs in function of receptors for the key monoamine neurotransmitters. Such wrong receptor function may be caused by depletion of monoamine neurotransmitters, by abnormalities in the receptor, or by problems with signal transduction on postreceptor level.
The common final result of chronic treatment by majority of antidepressants is the down-regulation or up-regulation of postsynaptic or presynaptic receptors (Table 5.7). The delay of clinical response corresponds with these receptor alterations, hence many receptor hypotheses of affective disorders were formulated and tested.
Table 5.7. Effect of depression and antidepressant treatment on receptor sensitivity
|
system |
receptor system |
treatment by antidepressants |
depression |
|
adrenergic |
b1-AR (postsynaptic, exc.) |
¯ |
|
|
a2-AR (presynaptic, inh.) |
¯ |
|
|
|
a2-AR (postsynaptic, inh.) |
|
¯ |
|
|
a1-AR (postsynaptic, exc.) |
|
|
|
|
serotonergic |
5-HT2 (exc.) |
¯ |
|
|
5-HT1A (somatodendritic autoreceptors, inh.) |
¯ |
¯? |
|
|
5-HT1A (postsynaptic, inh.) |
|
¯ |
|
|
5-HT1B (terminal autoreceptors, inh.) |
¯ |
|
|
|
other |
ACh |
¯ |
|
|
GABA |
|
¯ |
|
|
DA |
|
¯ |
|
|
corticosteroid receptors |
|
¯ |
ACh - acetylcholine, GABA - g-amino butyric acid, DA - dopamine, AR – adrenoceptors.
- increased, ¯ ‑ decreased
When activated by catecholamines, the presynaptic a2-adrenergic receptors can inhibit NE or 5-HT release. Some antidepressants reduce sensitivity of brain a2‑adrenoreceptors. Hypothesis about role of presynaptic a2-AR was formulated:
There is increased density of presynaptic a2-adrenergic receptors in high affinity state in depression.
Several lines of evidence indicate that an enhancement of 5-HT neurotransmission might underlie the therapeutic response to different types of antidepressant treatment, so, general serotonin receptor hypothesis was suggested:
Depression is connected with these abnormalities in serotonin receptors:
5-HT2 up-regulation
5-HT1A desensitisation
Abnormal signal transduction following 5-HT binding to receptor
Very elegant but not confirmed was receptor catecholamine hypothesis:
Supersensitivity of catecholamine receptors in the presence of low levels of serotonin is the biochemical basis of depression.
The common result of chronic treatment by majority of antidepressants is the down regulation of b-adrenergic receptors, which can be modulated by interaction with the 5-HT system, DA system, neuropeptides and hormones. Classical norepinephrine receptor hypothesis of affective disorders was formulated (Figure 5.1):
There is increased density of postsynaptic b-AR in depression (due to decreased NE release, disturbed interactions of noradrenergic, serotonergic and dopaminergic systems, etc.). Long-term antidepressant treatment causes down regulation of b1-AR (by inhibition of NE reuptake, stimulation or blockade of receptors, regulation through serotonergic or dopaminergic systems, etc.). Transient increase of neurotransmitter availability can cause fault to mania.
Apparently paradoxical increase of intracellular cAMP levels were observed at decreased density of b-AR, so increased cAMP system activity seems to be fundamental in therapeutic action of antidepressants. So, postreceptor hypotheses of affective disorders are tested at present time.
Figure 5.1. Classical noradrenergic receptor hypothesis
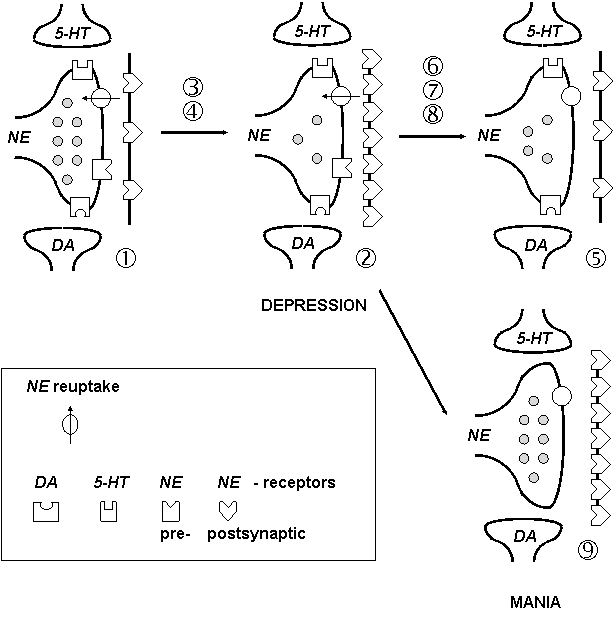
(See: Lopez D. et al.: The Essential Brain. Current Topics Sci. Med. Merck 1991.)
5.5. Postreceptor hypotheses
The introduction of new, more selective antidepressants led to new reflection upon the mechanism of their action. Most promising are recent investigations of the second messenger systems, the adenylyl cyclase system, the phosphatidylinositol system, G proteins, transcription factors etc. Modulation of receptor sensitivity, either down‑regulation or up‑regulation, is most probably dependent on the second and third messenger systems. Selected postreceptor hypotheses are shown in Table 5.8.
Table 5.8. Postreceptor hypotheses
|
Second messenger dysbalance hypothesis: |
|
Affective disorders arise from the dysbalance of the two major intraneuronal signal-amplification systems, with depression resulting from a hypofunction of the AC-cAMP kinases pathway together with dominance of the PLC-CaM/C kinases system, and mania resulting from the converse. |
|
G protein hypothesis: |
|
They are changes either density or function of subunits of G proteins during depression. (e.g. increase of Gs protein) |
|
Molecular model of affective disorders: |
|
Clinical heterogeneity observed at depressed patients can be explained by changes in signal transduction pathways which regulate two or more different neurotransmitter systems and which affect the neuron activity. G proteins, phosphatases and transcription factors are studied mainly. |
|
Molecular and cellular theory of depression: |
|
Transcription factor, cAMP response element-binding protein (CREB), is one intracellular target of long-term antidepressant treatment and brain-derived neurotrophic factor (BDNF) is one target gene of CREB. Chronic stress leads to decrease in expression of BDNF in hippocampus. Long-term increase in levels of glucocorticoids, ischemia, neurotoxins, hypoglycaemia etc. decreases neuron survival. Long-term antidepressant treatment leads to increase in expression of BDNF and his receptor trkB through elevated function of serotonin and norepinephrine systems. |
5.6. Molecular and cellular theory of depression
Recent studies have begun to characterize the action of stress and antidepressant treatments on signal transduction at postreceptor level. Long-term antidepressant treatments result in the sustained activation of the cAMP system in specific brain regions, including the increased function and expression of the transcription factor cAMP response element-binding protein (CREB). The activated cAMP system leads to the regulation of specific target genes, including the increased expression of brain-derived neurotrophic factor (BDNF) in hippocampus and cortex.
It was found that stress can decrease the expression of BDNF and lead to atrophy of the same populations of stress-vulnerable hippocampal neurons. Decreased size and impaired function of these neurons may be involved in depression; this possibility is supported by clinical imaging studies, which demonstrate a decreased volume of certain brain structures. These findings constitute the framework for a molecular and cellular hypothesis of depression (Table 5.8), which assume that stress-induced vulnerability and therapeutic action of antidepressant treatments occur via intracellular mechanisms that decrease or increase, respectively, neurotrophic factors necessary for the survival and function of particular neurons. This hypothesis also explains how stress and other types of neuronal insult can lead to depression in vulnerable individuals.
5.7. Molecular mechanism of action of antidepressants
A model for the molecular mechanism of action of antidepressant treatments is shown on Figure 5.2. Antidepressant treatment causes inhibition of serotonin and norepinephrine reuptake or breakdown. Short-term antidepressant treatment increase extracellular levels of serotonin and norepinephrine. Long-term treatment leads to decrease in the function and expression of serotonin and norepinephrine receptors, to increase in the cAMP signal transduction and to increase in expression of CREB. Increased activity of the cAMP signal transduction cascade indicates that the functional output of 5-HT and NE are up-regulated, even though levels of certain 5-HT and NE receptors are down-regulated. Expression of BDNF and its receptor trkB is also increased by long-term antidepressant treatment, so increased neuronal survival, function, and remodelling of synaptic architecture are provided.
Figure 5.2. A model for the molecular mechanism of action of antidepressant treatments

(See: Duman et al.: A Molecular and Cellular Theory of Depression. Arch. Gen. Psychiatry, 1997; 54: 597-606.)