Schizophrenia is specific human disease. It is group of diseases characterized by delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behaviour, of negative symptoms (Table 4.1). Symptoms of schizophrenia can be subcategorized into:
- Positive symptoms
- Negative symptoms
- Cognitive symptoms
- Aggressive/hostile symptoms
- Depressive/anxious symptoms
In this chapter, environmental, genetic, neurodevelopmental and biochemical hypotheses of schizophrenia are presented. Classification, clinical description and criteria for diagnosis of schizophrenia are not described.
For practical purposes the descriptive psychopathology of schizophrenia can be treated in three sections:
- Purely positive symptoms (hallucinations and other abnormal experiences, delusions and catatonia)
- What used to be referred to as psychological deficit, purely negative symptoms – impaired attention, intelligence, memory, perception and will.
- Traditional psychopathological groupings containing positive and negative symptoms (mixed) – thought disorder and disturbances of emotions.
Table 4.1. Positive and negative symptoms of schizophrenia
|
negative |
positive |
|
alogia |
hallucination |
|
affective flattening |
delusions |
|
avolition – apathy |
bizarre behaviour |
|
anhedonia – asociality |
positive formal thought disorder |
|
attentional impairment |
|
Biological models of schizophrenia can be divided into three related classes:
- Environmental models
- Genetic models
- Neurodevelopmental models
4.1. Environmental models
Environmental models suppose that either stress or physical factors are the main evoking phenomenon in schizophrenia (Table 4.2).
Table 4.2. Environmental models of schizophrenia
|
Model of “evocative influence of complex social demands”: |
|
There are four criteria for schizophrenia-evoking stress:
Most stresses of this nature will be non-pathogenic; a schizophrenia-evoking effect occurs only in conjunction with a specific genetic liability. |
|
Non-psychosocial environmental model: |
|
An unspecified number of varieties of physical factors will be sufficient to produce the specific cerebral lesions and dysfunction thought to be characteristic of schizophrenia, regardless of the presence or absence of a genetic susceptibility (see “Neurodevelopmental models” below). |
(See: Jablensky A.: Schizophrenia: the epidemiological horizon. In: Schizophrenia, Hirsch S.R. and Weinberger D.R., eds., Blackwell Science, pp. 206-252, 1995.)
4.2. Genetic models
Evidence for a genetic contribution to schizophrenia comes from twin and familiar studies. The genetic factors in schizophrenia have specificity as they do not increase the risk for major affective disorders or delusional disorder. Clearly, schizophrenia is clinically or phenotypically heterogeneous, but whether this variety is paralleled by etiological heterogeneity or to what extent is problematic. Genetic models are summarized in Table 4.3. Published data indicates that monogenic models could be rejected and multifactorial threshold model or mixed models are favoured.
Table 4.3 Genetic models
|
Distinct heterogeneity model: |
|
Schizophrenia is a collection of several separate diseases, each associated with single major locus (SML) that may be inherited either dominantly or recessively. In addition, there are sporadic, environmentally caused cases. |
|
Monogenic models (single major locus models): |
|
Schizophrenia might be a single-gene dominant disorder with highly variable expression or reduced penetrance of the trait; i.e. all cases of schizophrenia share the same single major locus (SML). |
|
Multifactorial-polygenic threshold model: |
|
Schizophrenia is the result of a combined effect of multiple genes interacting with variety of environmental factors; i.e. several or many genes, each of small effect, combine additively with the effects of non-inherited factors. The liability to schizophrenia is linked to one end of the distribution of a continuous trait, and there may be a threshold for the clinical expression of the disease. |
|
A mixed or combined model: |
|
The model includes the elements of some, or all, of the above three. |
(See: Jablensky A.: Schizophrenia: the epidemiological horizon. In: Schizophrenia, Hirsch S.R. and Weinberger D.R., eds., Blackwell Science, pp. 206-252, 1995; Asherson P., Mant R., McGuffin P.: Genetics and schizophrenia. In: Schizophrenia, Hirsch S.R. and Weinberger D.R., eds., Blackwell Science, pp. 253-274, 1995.)
4.3. Neurodevelopmental models
The leading hypothesis for the aetiology of schizophrenia is related to disturbance in normal brain development. The principal assumption is that normal brain development is disrupted in specific ways at critical periods and the resulting lesion produces the symptoms of schizophrenia only through interaction with the normal maturation processes in the brain, which occur in late adolescence or early adulthood.
Neurodevelopmental hypothesis states that:
A substantial group of patients, who receive diagnosis of schizophrenia in adult life, have experienced a disturbance of the orderly development of the brain decades before the symptomatic phase of the illness.
The neurodevelopmental model therefore directs attention to both genetic and no genetic risk factors that may have impacted on the developing brain during prenatal and perinatal life; pregnancy and birth complications (PBCs) are considered in psychiatry:
- viral infections in utero
- gluten sensitivity
- brain malformations
- obstetric complications
Genetic contribution to schizophrenia development consists in wrong genetic program for the normal formation of synapses and migration of neurons in the developing brain. These risk factors may have the final common effect on nerve growth factors reduction resulting in structural abnormalities, selection of wrong neurons to survive in the fetal brain, neuron migration to the wrong places, neuron inervation of wrong targets or mix-up of the nurturing signals.
4.4. Delayed onset of symptoms
We can propound the question, why is the illness manifestation delayed typically for about two decades after birth? Delayed onset of symptoms of schizophrenia (in early adult) can be explained by different mechanisms:
- There is an additional pathological process occurring around the time of onset of the clinical symptoms
- An interaction between a static developmental defect and normal developmental events that occur in early adulthood is necessary
It is supposed that onset of schizophrenia can be initiated by wrong organization, elimination and restructuring of synapses during adolescence, which may be or not secondary to the maldevelopment in utero.
4.5. Neurodegenerative hypothesis
Recent post mortem studies on schizophrenics described a multitude of morphological changes in different brain structures. The most often reported structural alterations are in the limbic system, mainly in hippocampal formation and parahippocampal and cingulate gyri. However, other structures such as the thalamus, frontal and temporal cortex and basal ganglia seem to be affected too.
Neurodegenerative hypothesis of schizophrenia suggest an ongoing neurodegenerative processes with loss of neuronal function during the course of the disease. Excitotoxic hypothesis proposes that neurons degenerate as a consequence of excessive glutamatergic neurotransmission.
Combined neurodevelopmental/neurodegenerative hypothesis suggest that schizophrenia may be a neurodegenerative process superimposed on a neurodevelopmental abnormality.
4.6. Biochemical basis of schizophrenia
The biochemical hypotheses of schizophrenia are orientated towards the role of neurotransmitters and their receptors; dopamine, serotonin, glutamate, GABA, norepinephrine and so on are considered (Table 4.4). Dopamine plays a key role in biochemical hypotheses of schizophrenia. Dopamine hypothesis of schizophrenia was formulated almost 40 years ago by Randrup and Munkvad (1965) and plays a prominent role in schizophrenia research hitherto. Basis or motives for dopamine hypothesis are summarized in Table 4.5.
|
According to the classical dopamine hypothesis of schizophrenia, psychotic symptoms are related to dopaminergic hyperactivity in the brain. Hyperactivity of dopaminergic systems during schizophrenia is result of increased sensitivity and density of dopamine D2 receptors. This increased activity can be localized in specific brain regions. |
Table 4.4. Biochemical hypotheses of schizophrenia
|
classical dopamine |
GABAergic |
|
norepinephrine |
glutamatergic |
|
serotonin |
peptidergic |
|
monoaminoxidase |
membrane |
|
revisited dopamine |
transmethylation |
Table 4.5. Basis of classical dopamine hypothesis of schizophrenia
|
Dopamine-releasing drugs (amphetamine, mescaline, diethyl amide of lysergic acid - LSD) can induce state closely resembling paranoid schizophrenia. |
|
Conventional neuroleptic drugs, that are effective in the treatment of schizophrenia, have in common the ability to inhibit the dopaminergic system by blocking action of dopamine in the brain. |
|
Neuroleptics raise dopamine turnover as a result of blockade of postsynaptic dopamine receptors or as a result of desensitisation of inhibitory dopamine autoreceptors localized on cell bodies. |
Revised dopamine hypothesis of schizophrenia postulate that a reduced striatal inhibition on the thalamus, caused by either an increased dopaminergic or a reduced glutamatergic tone, should lead to an increase in arousal and psychomotor activity and to an increased sensory input transmitted to the cortex. If a certain threshold is exceeded the integrative capacity of the cortex will become insufficient and this will lead to positive symptoms of schizophrenia. An excessive dopaminergic function may also lead to a disintegration of motor functions. This inhibitory function of the striatum appears to be exerted via the indirect pathways (Figure 4.1 A). The direct pathways (Figure 4.1 B) should be able to mediate the excitatory glutamatergic input from the cortex to the thalamus; the dopamine input in the direct pathways appears to be excitatory, and dopamine should thus be behaviourally stimulating also via the direct pathways.
Hypothetical scheme of interactions, leading to psychotogenic responses, is shown in Figure 4.2.
Figure 4.1. Excitatory and inhibitory influence of dopamine on the direct and indirect pathways
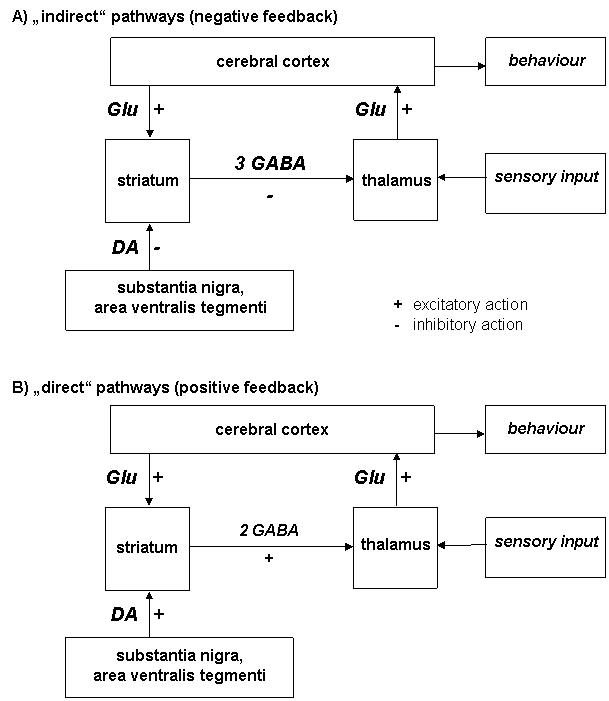
Glu – glutamate, DA – dopamine, GABA - g-amino butyric acid
(See: Carlsson A.: The dopamine theory revisited. In: Schizophrenia, Hirsch S.R. and Weinberger D.R., eds., Blackwell Science, pp. 379-400, 1995.)
Figure 4.2. Potential psychogenic pathways and sites of action of psychotogenic and antipsychotic agents
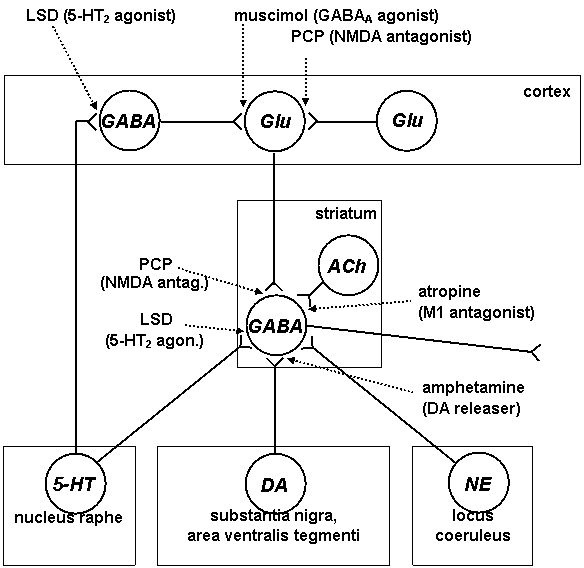
LSD – diethyl amid of lysergic acid, PCP – phencyclidine, 5-HT – serotonin, Glu – glutamate, DA – dopamine, GABA – g-amino butyric acid, ACh – acetylcholine, NE – norepinephrine, NMDA – N-methyl-D-aspartate
(See: Carlsson A.: The dopamine theory revisited. In: Schizophrenia, Hirsch S.R. and Weinberger D.R., eds., Blackwell Science, pp. 379-400, 1995.)